- HCN-Kanal
-
HCN-Kanal —
Sekundär- bis Quartärstruktur Heteromultimer, multi-pass Membranprotein Bezeichner Gen-Name(n) HCN1, HCN2, HCN3, HCN4 Transporter-Klassifikation TCDB 1.A.1 Bezeichnung Spannungsgesteuerte Ionenkanäle HCN-Kanäle (aus dem Englischen: hyperpolarization-activated, cyclic nucleotide-gated cation channel) stellen eine kleine Unterfamilie der Proteingruppe der Ionenkanäle dar. Sie sind eine Untergruppe der zyklonukleotid-regulierten Kationenkanäle, die ihrerseits zur Familie der Porenschleifen-Kationenkanäle gehören.[1]
Derzeit sind vier HCN-Untereinheiten (HCN1 bis HCN4) bekannt, die beim Menschen hauptsächlich in Herz und Gehirn exprimiert werden. Der von HCN-Kanälen getragene Strom wird in vielen Fällen auch als ‚Schrittmacher-Strom‘ bezeichnet, da er an der Kontrolle des Herzrhythmus beteiligt ist und in spontan aktiven Nervenzellen rhythmische Aktivität fördert. Er wird entweder als Ih (für engl. hyperpolarization) oder If (für engl. funny) bezeichnet.
Topologie von HCN-Kanälen
Der generelle Aufbau eines HCN-Kanalkomplexes ist stets gleich: Der Komplex wird durch Aneinanderlagern von vier einzelnen HCN-Proteinen (Untereinheiten ) gebildet, welche in vivo vier verschiedene Homotetramere bilden, die sich in ihren biophysikalischen Eigenschaften voneinander unterscheiden. Die Untereinheiten bilden aneinander gelagert eine zentrale Pore, die den Durchtritt von Ionen ermöglicht. Jede Untereinheit besteht aus drei wichtigen Strukturen: dem zytosolischen N-Terminus, dem zytosolischen C-Terminus mit der Zyklonukleotidbindedomäne und die Transmembrandomäne, welche die Pore beinhaltet und mechanistisch an der Kanalöffnung beteiligt ist. (siehe Abbildung)
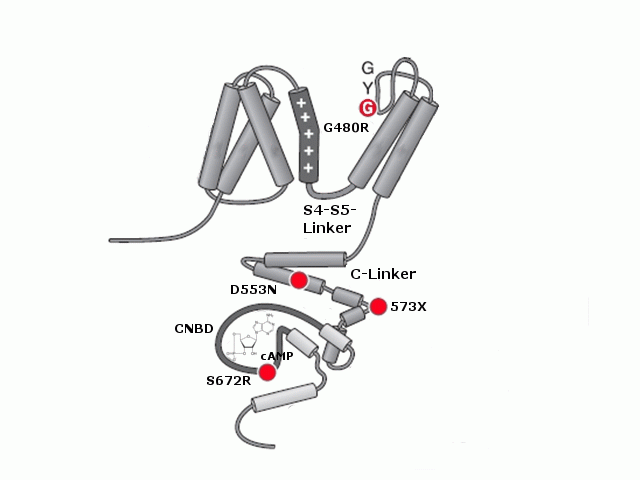
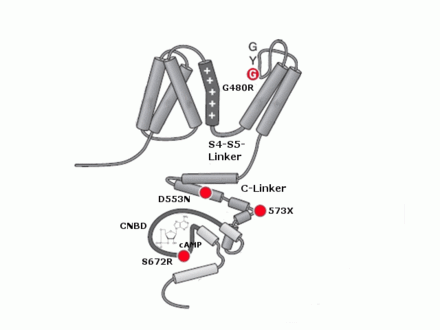 Struktur eines HCN-Kanals, abgebildet ist eine Untereinheit mit sechs Transmembrandomänen und dem intrazellulären C-Terminus. Das S4-Segment enthält den Spannungssensor. Bekannte pathophysiologisch relevante Mutationen werden mit roten Punkten und der betroffenen Aminosäure dargestellt.
Struktur eines HCN-Kanals, abgebildet ist eine Untereinheit mit sechs Transmembrandomänen und dem intrazellulären C-Terminus. Das S4-Segment enthält den Spannungssensor. Bekannte pathophysiologisch relevante Mutationen werden mit roten Punkten und der betroffenen Aminosäure dargestellt.
Zyklonukleotidbindedomäne
Die Zyklonukleotidbindedomäne (CNBD, cyclic nucleotide binding domain) hat einen modulierenden Effekt auf die Kanalöffnung, abhängig davon, ob cyclisches Adenosinmonophosphat (cAMP) gebunden ist oder nicht.[2] Sie ist über den sogenannten C-Linker mit der Transmembrandomäne verbunden. Es gibt bereits Kristallstrukturen, die CNDBs mit gebundenem cAMP zeigen.[3] Die Tasche, in der das cAMP bindet, besitzt sieben Aminosäurereste, die mit dem Liganden interagieren.[4] Diese Aminosäurereste weisen unterschiedliche Aufgaben auf. So vermittelt der Rest R632 die Wirksamkeit einer cAMP-Bindung auf die Kanalöffnung.[4]
Die Bindung von cAMP an den Kanal führt zu einer verstärkten Kanalaktivität, da durch die geringe Konformationsänderung bei der Bindung eine anhaltende Hemmung (tonische Inhibition) aufgehoben wird.[5][6] Nach einem Modell[7] wirkt der in Abwesenheit von cAMP kompakte C-Linker inhibitorisch auf die Kanalöffnung. Bindet cAMP an die Zyklonukleotidbindedomäne, führt das zu einer Änderung der Konformation des C-Terminus. Diese Änderung resultiert in verminderter Inhibition und destabilisiert den geschlossenen Zustand. Folglich erhöht cAMP die Öffnungswahrscheinlichkeit der HCN-Kanäle. Dadurch verschiebt sich die Spannungsabhängigkeit der Aktivierung. Dies bedeutet, dass sich die Kanäle bei gegebener cAMP-Konzentration so verhalten, als wären sie einer stärkeren Hyperpolarisation ausgesetzt. Somit besteht ein „höherer Druck“ sich zu öffnen.
Transmembrandomäne
Der transmembranäre Anteil einer HCN-Untereinheit besteht aus sechs alpha-helikalen Segmenten (S1–S6) und einer ionenleitenden Porenschleife zwischen S5 und S6. Ein hochkonservierter Asparaginrest in der extrazellulären Schleife zwischen S5 und der Porenschleife ist glykosyliert. Diese Modifikation ist essentiell für eine normale Oberflächenexpression der HCN-Kanäle.[8] Der Spannungssensor der HCN-Kanäle liegt im S4-Segment. Er besteht aus neun Arginin- oder Lysinresten, die an jeder dritten Position im S4-Segment lokalisiert sind.[9] Positiv geladene S4-Segmente finden sich in allen spannungsabhängigen Kationenkanälen mit 6-Transmembrandomänen-Topologie. HCN-Kanäle bilden hier jedoch eine Besonderheit. Eine Einwärtsbewegung des Segments durch die Ebene der Zellmembran als Antwort auf eine Hyperpolarisation führt zu einer Kanalöffnung, während sie bei allen anderen Kanälen zu einer Schließung führt.[10] Dieser Effekt ist Gegenstand aktueller Forschung. Erste Hinweise deuten auf die Verbindung von S4 zu S5 als molekulares Korrelat dieser andersartigen Antwort auf Spannung hin.[11][12]
Heteromerisierung
Es konnte zudem gezeigt werden, dass die Isoformen HCN1 und HCN2 miteinander koassemblieren und Heteromultimere bilden. Die Expression von Konkatameren (Wiederholungssequenzen einer DNA-Kette) von HCN1- und HCN2-Untereinheiten bei Mäusen zeigt, dass die Kinetik des entstehenden Kanals intermediär zwischen denen der einzelnen Isoformen liegt.[9][13] Diese Resultate verdeutlichen, dass eine Entstehung von Heteromultimeren in Zellen, die beide Isoformen exprimieren, möglich ist. Interessant an den Heteromultimeren ist, dass jede Untereinheit eine spezifische Charakteristik auf den Kanal überträgt. So lag die halbmaximale Aktivierung nahe der von HCN2-Homomultimeren, die Geschwindigkeit der Aktivierung entsprach hingegen der eines HCN1-Homomultimers. Die Reaktion der HCN1/HCN2-Kanäle auf cAMP war sogar exakt die gleiche wie die von HCN2-Kanälen. Eine Heteromultimerisierung anderer Stöchiometrie mit variierenden Isoformen ist demnach im nativen Gewebe ebenfalls denkbar. Weitere Experimente lassen zudem vermuten, dass native HCN-Kanäle β-Untereinheiten enthalten können.[14] Die Koexpression des Proteins MinK-related peptide 1 (MIRP1) mit HCN1 oder HCN2 erhöhte die Stromamplitude signifikant. MIRP1 könnte entweder die Oberflächenexpression verbessern oder den Kanalkomplex stabilisieren. Zudem nimmt MIRP1 Einfluss auf die Aktivierungs- und Deaktivierungskinetik der HCN-Isoformen, woraus sich die Variabilität der Kinetik in verschiedenen Studien erklären könnte. Allerdings konnte der Effekt von MIRP1 mit HCN-Kanälen nicht von allen Gruppen reproduziert werden. Daher ist eine Interaktion dieses KCNE-Proteins mit HCN unsicher.
Biophysik der HCN-Kanäle
Spannungsabhängigkeit der Aktivierung
Die Spannungsabhängigkeit der Aktivierung ist für die verschiedenen HCN-Kanäle unterschiedlich. Die ungefähre Spannung der halbmaximalen Aktivierung beträgt für HCN1 -70 mV, für HCN2 -95 mV, für HCN3 liegt sie im Bereich von -77 mV bis -95 mV und für HCN4 beträgt sie -100 mV.[15][16] Im Jahr 2005 konnte ein weiterer spannungsabhängiger Effekt gezeigt werden[17]. Sowohl der HCN-Kanal des Seeigelspermiums (spHCN), als auch der HCN1-Kanal der Säugetiere, können zwischen zwei verschiedenen Modi wechseln, abhängig von der vorhergegangenen Aktivität. Im Modus 1 öffnet der Kanal bei sehr negativen Spannungen, während im Modus 2 diese Öffnung um 50 mV ins Positive verschoben ist, also früher erfolgt. Im geöffneten Zustand wechselt der Kanal von Modus 1 in Modus 2, umgekehrt ist es im geschlossenen Zustand. Auch für den HCN2-Kanal konnte dieser Hystereseeffekt beobachtet werden, wenn auch weit schwächer als für den HCN1-Kanal. Für den HCN4-Kanal ließ sich der Effekt nicht beobachten.[18] Dieser Moduswechsel führt zu einer Spannungshysterese, mit funktioneller Relevanz für das aktivierungsabhängige „Kurzzeitgedächtnis“ des HCN-Kanals.[19]
Aktivierungskinetik
Die Kinetik der Aktivierung ist ebenso wie die Spannungsabhängigkeit für die verschiedenen Isoformen unterschiedlich. HCN1 ist der Kanal mit der schnellsten Öffnungskinetik mit einem tau-Wert von 25 bis 300 ms abhängig von der Spannung.[20] Der HCN4-Kanal hat die langsamste Öffnungskinetik mit einem tau-Wert, der zwischen einigen hundert Millisekunden bis zu mehreren Sekunden beträgt. Die Kinetiken von HCN2 und HCN3 liegen zwischen denen von HCN1 und HCN4.[21] Die Spannungsabhängigkeit und die Aktivierungskinetik der HCN-Kanäle werden sehr stark durch experimentelle Parameter (z.B. Messprotokoll, Temperatur) und das intrazelluläre Milieu beeinflusst. Diese Eigenschaft der HCN-Kanäle könnte die Variabilität der biophysikalischen Parameter erklären, die verschiedene Gruppen beobachten konnten.[22]
Einzelkanalleitfähigkeit
Die Einzelkanalleitfähigkeit der HCN-Kanäle wird kontrovers diskutiert. Die erstmalig beschriebene Einzelkanalleitfähigkeit, die sich durch neuere Messungen bestätigen ließ, lag im Bereich von 1 pS und war damit sehr klein.[23] Es wurden jedoch auch bis zu 30 mal größere Einzelkanalleitfähigkeiten publiziert.[24] Der beobachtete Unterschied mag in den unterschiedlichen Messkonfigurationen begründet sein, könnte jedoch auch durch in-vivo-Regulationsmechanismen dynamisch verändert werden. Ebenfalls sollte beachtet werden, dass die Einzelkanäle teilweise nicht der Kinetik entsprachen, die man von HCN-Kanälen erwartet.[24] Daher bleibt eine endgültige Bestimmung der Einzelkanalleitfähigkeit abzuwarten.
Ionenselektivität
HCN-Kanäle leiten sowohl Natrium- als auch Kaliumionen im Verhältnis 1:4. Sie werden von millimolaren Konzentrationen Caesium geblockt[25] Es wurde zudem eine geringe Leitfähigkeit für Calcium beschrieben, deren funktionelle Relevanz derzeit nicht geklärt ist.[26][27]
Obwohl HCN-Kanäle auch Natrium leiten, tragen sie das für Kaliumkanäle typische GYG-Motiv, das den Selektivitätsfilter bildet. Dies ließ zunächst vermuten, dass der Kanal selektiv nur Kalium durchlässt. Bis jetzt ist unklar, wie der Kanal beide Ionensorten leitet. Allerdings wird vermutet, dass die GYG-Motive der einzelnen Untereinheiten in größerem Abstand zueinander liegen als in „reinen“ Kaliumkanälen und so den Durchtritt des größeren Natriums ermöglichen. Die extrazelluläte Kaliumkonzentration hat großen Einfluss auf die Stromamplitude, aber auch auf das Verhältnis der Leitfähigkeiten für Natrium und Kalium. Dabei führt ein Anstieg der extrazellulären Kaliumkonzentration zu einer vergrößerten Stromamplitude bei einem etwas geringeren Verhältnis von Kalium- zu Natriumleitfähigkeit.[28][29]
In Abwesenheit von Kalium leiten HCN-Kanäle kaum noch Natrium.[30] HCN-Kanäle leiten keine Anionen, dennoch reagieren sie auf eine Änderung der extrazellulären Chloridkonzentration.[31]
Modulation durch cAMP
Obwohl alle vier HCN-Untereinheiten hochkonservierte C-Linker und Zyklonukleotid-Bindestellen besitzen, fällt die Modulation durch cAMP unterschiedlich stark aus. Die Spannungsabhängigkeit der Aktivierung von HCN2 und HCN4 werden durch Gabe von cAMP um +10 bis +25 mV verschoben.[15][21][32] Im Gegensatz dazu sind HCN1 und HCN3 nur schwach durch cAMP reguliert.[15][16][32] Ersetzt man stattdessen die Zyklonukleotidbindestelle von HCN4 durch die von HCN3, so bleibt die cAMP-Sensitivität vollständig erhalten.[16] Die Bindestelle von HCN3 ist also in der Lage cAMP zu binden. Allerdings scheint eine Änderung in der Untereinheitenstruktur von HCN3 dafür zu sorgen, dass die Zyklonukleotidbindestelle funktionell stillgelegt wird.
Expression von HCN-Kanälen
Die Expression der verschiedenen HCN-Kanäle beschränkt sich größtenteils auf Herzmuskel- und Nervengewebe. HCN1 wird im Gehirn stark exprimiert, wohingegen im Herzen HCN2 und HCN4 vermehrt vorliegen. Für den HCN1-Kanal findet man eine starke Expression im Gehirn. Dort findet sich der Kanal vor allem im Neocortex, der CA1-Region des Hippocampus, in den Colliculi superiores und im Stratum moleculare (Molekularschicht) des Kleinhirns.[33][34] Eine Arbeitsgruppe konnte eine robuste Expression von HCN1-Kanälen im Sinusknoten und den Purkinje-Fasern des Kaninchen-Herzens nachweisen, allerdings ist diese Herzregion stark innerviert und eine Kontamination mit neuronalen Zellen kann nicht ausgeschlossen werden.[35] Im menschlichen Sinusknoten konnte kürzlich gezeigt werden, dass der HCN1-Kanal mit dem HCN4-Kanal koexprimiert.[36]
Der HCN2-Kanal findet sich ebenfalls in zahlreichen Gebieten des Gehirns, so im Riechkolben (Bulbus olfactorius), im Hippocampus, im Thalamus und in der Amygdala.[34] Im Herzen kann der Kanal sowohl im Ventrikel, als auch im Atrium (Vorhof) nachgewiesen werden.[21] Auch im Sinusknoten findet sich der HCN2-Kanal in einigen Spezies wie Ratte und Maus.[2] Der HCN3-Kanal ist – auf den gesamten Organismus bezogen – die am wenigsten exprimierte Isoform. Im Nervengewebe findet er sich sehr schwach exprimiert im Riechkolben, ansonsten sind HCN3-Signale kaum stärker als das Hintergrundsignal der in-situ-Hybridisierungen.[34] Im Herzen findet sich der Kanal nur in sehr wenigen Zellen des Reizleitungssystems.[2] Zusätzlich nimmt der HCN3-Kanal im Hinblick auf die Gewebeexpression eine besondere Rolle ein. Er konnte neben Nerven- und Herzmuskelgewebe als einzige HCN-Isoform in Niere und Leber nachgewiesen werden. Über seine Funktion in diesen Geweben gibt es bisher keine Daten.[20] Es lässt sich eine massive Expression von HCN4 in verschiedenen thalamischen Neuronen und in der Mitralzellregion des Riechkolbens zeigen.[34][37] In allen bisher untersuchten Spezies findet sich der HCN4-Kanal im Sinusknoten als dominante Isoform. Er macht dort circa 80 % der HCN-Expression aus.[35] Der verbleibende Anteil variiert von Spezies zu Spezies und wird entweder von HCN1 dominiert (im Kaninchen und Menschen)[35][36] oder von HCN2 (in der Maus)[38]. Auch in anderen Teilen des Reizleitungssystems (AV-Knoten, Purkinje-Fasern) überwiegt die HCN4-Expression. In Herzmuskelzellen der Maus hingegen spielt HCN4 eine weniger wichtige Rolle, dort dominiert HCN2.[21]
Regulation von HCN-Kanälen
HCN-Kanäle werden sowohl durch extra- wie auch intrazelluläre Mechanismen sehr intensiv reguliert. Dabei werden die Oberflächenexpression, die Lokalisation in bestimmte Kompartimente der Zelle und die funktionellen Eigenschaften des Kanals beeinflusst. Die Regulation kann sowohl durch niedermolekulare Substanzen geschehen, wie auch durch Interaktion mit anderen Proteinen.
Interaktion mit niedermolekularen Substanzen
Interaktion mit Ionen
Es ist bekannt das sowohl Chlorid-Ionen, als auch Protonen regulierend auf HCN-Kanäle wirken können. Wie bereits erwähnt sind HCN-Kanäle reine Kationenkanäle. Allerdings wird ihre Leitfähigkeit beeinflusst durch die Konzentration an freien Chloridionen.[31] Ersetzt man extrazelluläres Chlorid durch Anionen die größer sind, so wird der durch HCN-Kanäle getragene Strom kleiner. Diese Sensitivität wird vermittelt durch einen Arginin-Rest der sich nur bei HCN2 und HCN4 findet, nicht jedoch bei HCN1.[39] Daher ist auch nur bei diesen Isoformen der Effekt ausgeprägt zu beobachten. Eine Regulation durch Chlorid ist möglicherweise relevant für physiologische Prozesse im Herzen.
Protonen können HCN-Kanäle sowohl intra- als auch extrazellulär regulieren.[40] [41]. Ein einzelner Histidinrest im HCN2 der MAus ist dabei für die intrazelluläre Reaktion auf geänderte pH-Werte verantwortlich. Durch Änderungen des pH-Wertes verschiebt sich das Potential der halbmaximalen Aktivierung bei saurem pH-Wert zu mehr hyperpolarisierten Werten, bei alkalischem pH-Wert zu mehr depolarisierten Werten im Vergleich zum physiologischen pH-Wert von 7,4. Extrazellulärer pH hat einen Einfluss auf HCN1 und HCN4-Kanäle, die sich in Geschmackszellen von Ratten finden. Ein pH-Wert kleiner als 5,0 aktiviert dabei die Kanäle, indem er die Aktivierungskinetik beschleunigt und die Aktivierungsschwelle herabsetzt, so dass die Kanäle früher öffnen.[41]
Interaktion mit Phosphatidylinositol-4,5-bisphosphat (PIP2)
PIP2 ist ein allosterischer Aktivator von HCN2-Kanälen, der von der intrazellulären Seite an den Kanal bindet und die Kanal-Aktivierung erleichtert.[42] [43] Dies geschieht auch in der Abwesenheit von zyklischen Nukleotiden. Durch die Interaktion mit PIP2 gelangen HCN-Kanälen erst in einen physiologischen Öffnungsbereich. Außerdem kann durch Abbau von PIP2 die Kanalaktivität durch die Zelle reguliert werden und so an die Bedürfnisse der Zelle angepasst werden.
Interaktion mit anderen Proteinen
Interaktion mit Kinasen
Für die Proteinkinasen Src und p38-MAP ist eine Interaktion mit HCN-Kanälen beschrieben.[44][45] Die Tyrosin-Kinase Src bindet nachweislich an die Untereinheiten von HCN1[46], HCN2[47] und HCN4[48]. Eine Interaktion mit der Kinase führt zur Phosphorylierung eines Tyrosinrestes, der durch die gesamte HCN-Familie konserviert ist. Durch diese Phosphorylierung beschleunigt sich die Öffnung des Kanals und die Spannung der halbmaximalen Aktivierung verschiebt sich zu positiveren Potentialen. Die Regulation von HCN durch Src-Kinasen konnte bereits im Herzen von Mäusen und Ratten, sowie in Neuronen gezeigt werden[47][49].
Interaktion mit Transmembranproteinen
Wie viele andere Ionenkanäle auch, besteht der HCN-Kanal aus einem Transmembrankomplex, an den sich andere Transmembranproteine anlagern können, um mit dem Kanal zu interagieren. Für das Protein MiRP1 wurde eine Interaktion mit verschiedenen HCN-Kanälen publiziert.[14] [50] [51]. Das Protein besteht aus einer einzelnen Tranmembrandomäne, die sich an den Kanal anlagert. Dadurch wird die Stromdichte erhöht und die Kanalöffnung von HCN2 beschleunigt[14], die von HCN4 wird verlangsamt. Wie diese kontroverse Wirkung auf die eigentlich sehr ähnlich Untereinheiten übertragen wird, ist derzeit nicht bekannt. Ein weiteres Transmembranprotein, mit dem HCN-Untereinheiten interagieren können, ist KCR1.[52] Es verringert die Stromdichte von HCN2 und hat Einfluss auf die Einzelkanalleitfähigkeit. Auch hier ist der Mechanismus bisher nicht geklärt.
Physiologische Rolle der HCN-Kanäle
Rolle der HCN-Kanäle in Neuronen
In einigen Nervenzellen (Neuronen) des Zentralnervensystems wirken HCN-Kanäle als echte Schrittmacher. Am besten untersucht ist diese Funktion in Schaltneuronen des Thalamus. Hier verursacht der HCN-Kanal eine Depolarisation der ein Aktionspotential folgt, ausgelöst durch T-Typ Calciumkanäle. In Purkinjezellen des Kleinhirns sind HCN-Kanäle keine wirklichen Schrittmacher, sie verhindern jedoch nach einem Aktionspotential eine übermäßig lange Refraktärzeit, indem sie einer Hyperpolarisation entgegenwirken und die Zelle schnell wieder auf das ursprüngliche Membranpotential einstellen.[53]
Es wird außerdem vermutet, dass HCN-Kanäle an der Fortleitung und Integration erregender (exzitatorischer) synaptischer Einflüsse beteiligt sind. Der Strom der HCN-Kanäle funktioniert wie ein Kurzschluss, der eine übermäßig lange und starke Erregung verhindert.[54] [55] Blockiert man HCN-Kanäle, so kommt es zu verstärkter temporaler Summation. Es konnte zudem gezeigt werden, dass die Dichte der HCN-Kanäle mit der Entfernung vom Nervenzellkörper (Perikaryon) zunimmt. Dies beschleunigt distale Erregungen relativ zu proximalen und führt im Endeffekt zu einem identischen Zeitverlauf aller Erregungen eines Dendriten.
Erst im Jahr 2007 wurde ein Einfluss von HCN-Kanälen in der Kontrolle des räumlichen Gedächtnisses festgestellt.[56] Durch die Regulation der intrazellulären cAMP-Konzentration, die über α-Adrenorezeptoren gesenkt und über Dopaminrezeptoren erhöht wird, wird die Offenwahrscheinlichkeit der HCN-Kanäle moduliert. Durch eine Regulation der HCN-Kanäle wird in den Neuronen der Membranwiderstand angepasst. Dies kann eine Reaktion auf eingehende Informationen erleichtern oder erschweren. So werden unwichtige Informationen durch vorherige Aktivierung der HCN-Kanäle gefiltert und in Konzentrationsphasen wichtige Information leichter verarbeitet. Neben dem räumlichen Gedächtnis hat HCN1 auch eine Funktion für das Erlernen von motorischen Fähigkeiten. Eine Deletion von HCN1 verursacht starke Lerndefizite in diesem Bereich.[57]
Rolle der HCN-Kanäle im Herzen
Die bekannteste Funktion der HCN-Kanäle ist die Generierung eines kardialen Schrittmacherpotentials. Hier wirkt ihr Strom als Hauptkomponente der diastolischen Depolarisation im Sinusknoten. Auch die β-adrenerge Stimulation der Herzfrequenz erfolgt über die Wirkung von cAMP auf HCNKanäle.[58] Eine sympathische Stimulation aktiviert die Kanäle und beschleunigt dadurch den Herzschlag, vagale Stimulation verlangsamt den Herzrhythmus.
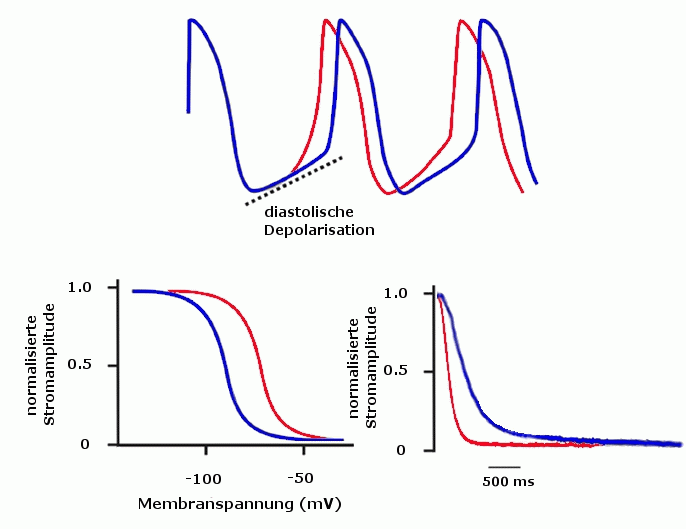
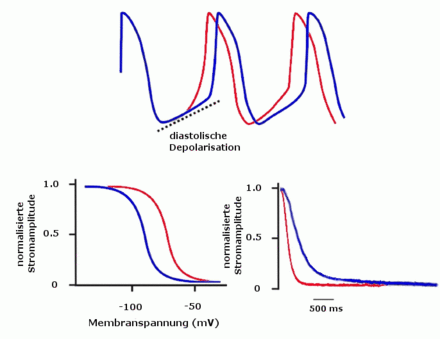 Die Rolle von HCN bei der Generierung des Herzrhythmus (oben) Schrittmacherfunktion durch diastolische Depolarisation (blau) und unter adrenerger Stimulation (rot) (unten) In der Anwesenheit von cAMP (rot) verschiebt sich die Aktivierungskurve (links), zudem öffnen HCN-Kanäle schneller (rechts)
Die Rolle von HCN bei der Generierung des Herzrhythmus (oben) Schrittmacherfunktion durch diastolische Depolarisation (blau) und unter adrenerger Stimulation (rot) (unten) In der Anwesenheit von cAMP (rot) verschiebt sich die Aktivierungskurve (links), zudem öffnen HCN-Kanäle schneller (rechts)HCN4 ist in der Entwicklung von Herzzellen von Mäusen von entscheidender Bedeutung. Ohne eine Expression von HCN4 entwickeln sich deren Schrittmacherzellen nicht zu adulten Zellen und die Tiere versterben noch im Mutterleib.
Knockout-Versuche
Zur Untersuchung der möglichen Funktionen der einzelnen HCN-Kanäle wurden verschiedene Knockout-Modelle der Maus erzeugt. Diese Modelle geben Aufschluss über physiologische und pathophysiologische Relevanz der HCN-Kanäle.
HCN1-Knockout
Ein globaler Gen-Knockout von HCN1 in Mäusen führt zu einem Defekt, der das Erlernen von motorischen Aufgaben betrifft.[59] Vor allem betroffen ist das Ausführen schneller Bewegungen. Dieser Effekt wird dem Verlust von HCN1 in den Purkinje-Neuronen des Kleinhirns zugeschrieben. Dort findet sich ein deutlicher Unterschied in Reaktion auf eine Membranhyperpolarisation. Die HCN1-defizienten Purkinje-Neurone bleiben wesentlicher länger im hyperpolarisierten Zustand und verringern ihre Aktionspotentialrate. Während bei Wildtyp-Purkinje-Neuronen die Feuerrate unabhängig vom Ausgangszustand ist, wirkt der Ausgangszustand der Erregung bei HCN1-defizienten Tieren auf die Feuerrate ein. Dieser Einfluss scheint das motorische Lernen zu behindern. In CA1-Neuronen hat der Verlust von HCN1 einen deutlich anderen Effekt. Hier verbessert er das räumliche Gedächtnis, sowohl kurz- als auch langfristig. Dieser Effekt wird durch eine verbesserte Langzeitpotenzierung an CA1-Neuronen vermittelt.[60]
HCN2-Knockout
In HCN2-defizienten Tieren lässt sich sowohl ein neuronaler, als auch ein kardialer Phänotyp beobachten.[38] Schon äußerlich unterscheiden sich die Tiere von ihren Wildtyp-Artgenossen durch verminderte Aktivität und Ataxie. Bei den Mutanten konnte elektrophysiologisch eine Absence-Epilepsie nachgewiesen werden. Die thalamokortikalen Neuronen reagieren hier vermutlich übermäßig stark auf Erregungen aus der Großhirnrinde. Zudem führt der Verlust von HCN2-Kanälen zu einem hyperpolarisierten Ruhemembranpotential, das die normalerweise inaktivierten T-Typ-Calciumkanäle aktivierbar macht und so zu möglichen Oszillationen führt. Der kardiale Phänotyp des HCN2-Knockout ist eine Sinusarrhythmie, charakterisiert durch variierende RR-Intervalle bei ansonsten normalem Elektrokardiogramm. Dieser Effekt beruht auf dem Verlust von HCN2-Kanälen in Herzmuskelzellen und ist kein sekundärer Effekt eines HCN2-Knockouts in Nervenzellen. Die Tatsache, dass die Mäuse weder bradykard sind, noch Probleme bei Belastung haben, deutet darauf hin, dass der HCN2-Kanal nicht für die Modulation der Herzfrequenz benötigt wird. Im Sinusknoten ist die Stromamplitude um 25 % reduziert. Zudem ist das maximale diastolische Potential 5 mV negativer als beim Wildtyp. Die somit fehlende Stabilisierung des Ruhemembranpotentials durch HCN2 führt zu einer Verzögerung des folgenden Aktionspotentials, sodass es zu Unregelmäßigkeiten im Herzrhythmus kommt.[60]
HCN4-Knockout
Ein globaler oder kardiospezifischer Verlust von HCN4-Kanälen führt zum Tod des Tieres in utero zwischen Tag 10 und 11,5.[61] Die Herzfrequenz der Tiere ist um 40 % geringer und es ist keine Reaktion auf cAMP zu beobachten. Der Verlust des HCN4-Kanals führt zum Verlust des Schrittmacherpotentials. Dies zeigt die Bedeutung von HCN4-Kanälen für die normale Funktion des Herzens auf. Unterdrückt man den HCN4-Kanal in adulten Tieren[62], so kommt es zu Sinuspausen. Die Tiere sind jedoch weder bradykard, noch zeigt sich eine fehlerhafte Modulation des Herzrhythmus. Daher scheint der HCN4-Kanal im adulten Tier weniger essentiell zu sein als in Embryonen.
Pathophysiologische Rolle der HCN-Kanäle
HCN-Kanäle haben vor allem in Herz und Gehirn physiologische Relevanz als primäre und sekundäre Schrittmacher. Daher sind diese Organsysteme auch pathophysiologisch von Änderungen an HCN-Kanälen betroffen. Die bisher in Folge von HCN-Mutationen beschriebenen Krankheitsbilder sind Epilepsien, Neuralgie und Herz-Arrhythmie.
Epilepsie
Absence-Epilepsien im Tiermodell
Ein direkter Effekt von Änderungen an HCN-Kanälen auf Epilepsien ist derzeit nur in Tiermodellen eindeutig nachgewiesen. Expemplarisch ist das Modell der Ratte vom Stamm WAG/Rij, das als Modell für Epilepsien vom Absence-Typ dient: Bereits im Jahr 1986 konnten für den Inzuchtstamm WAG/Rij-Wistar spike-wave discharges nachgewiesen werden, die von milden klinischen Symptomen begleitet wurden.[63] Diese sind typisch für Epilepsien vom Absence-Typ. Von ihrem Ausgangsstamm Wistar sind sie durch Verhaltensstudien nicht zu unterscheiden, sodass sich phänotypisch die Absence-ähnlichen Anfälle als einziges Differenzierungsmerkmal herausheben. Die Anfallsymptomatik ist der beim Menschen extrem ähnlich und unterscheidet sich nur in der Frequenz der Entladungen.[64] Alle anderen Symptome gleichen jedoch denen eines Anfalls beim Menschen. Die Generierung beidseitig (bilateral) synchroner spike-wave discharges ist nur in einem anatomisch und funktionell gesunden kortiko-thalamischen Netzwerk möglich, das sich zudem in einem begünstigenden Ausgangszustand befindet. Charakterisiert wird dieser durch eine leichte Hyperpolarisation der Pyramidenzellen der Großhirnrinde und der thalamischen Kerne, die sie anfällig machen für hochfrequente Aktionspotentialfolgen.
Eine genetische Komponente der Absence-Epilepsie ist sowohl beim Menschen als auch bei der Ratte so gut wie sicher.[65][66] Dies konnte für den Calcium-abhängigen Kaliumkanal[67] und den T-Typ Calciumkanal[68] sowie für die HCN-Kanäle HCN2[38] und HCN1 gezeigt werden. Die Bedeutung von HCN1-Strömen bei Absence-Epilepsien wird vor allem in zwei Publikationen deutlich. In der ersten Veröffentlichung[69] wurde der Strom von thalamokortikalen Neuronen in WAG/Rij-Ratten mit Cäsium oder dem HCN-Blocker ZD7288 blockiert, was zu einer verstärkten Entladung der Neuronen führte. Im Vergleich zum Wildtyp konnte eine Änderung des Potentials der halbmaximalen Aktivierung hin zu negativeren Potentialen gezeigt werden (-93,2 mV bei WAG-Tieren im Vergleich zu -88,0 mV im Wildtyp). Die WAG-Neuronen reagierten weniger gut auf cAMP. Zudem war die Expression der mRNA und des Proteins des HCN1-Kanals im Vergleich zum Wildtyp deutlich erhöht. Die Gruppe schließt daraus, dass der Anstieg der Expression mit der verminderten Reaktion auf cAMP für die überschießende Erregung thalamokortikaler Neurone in WAG-Tieren, und damit kausal für Absence-Anfälle, verantwortlich ist. Die zweite Veröffentlichung[70] beschreibt, dass ein rapider Verlust des HCN1-Transkripts dem ersten Anfall vorausgeht. Dieser Verlust wird hauptsächlich in apikalen Dendriten von Pyramidalzellen der Großhirnrinde beobachtet. Der unter anderem von HCN1-Kanälen getragene Ih-Strom ist in etwa halbiert. Dadurch wird die somatodendritische Kommunikation erleichtert und rückläufige Aktionspotentiale können schon in niedriger Frequenz einen Calcium-Einstrom bewirken. Es kommt in vielen dieser Neuronen zu einer intrinsisch hohen Feuerrate. Zusätzlich werden nach dem Verlust des HCN1-Kanals weitere Calciumkanäle rekrutiert, die für verstärkte Entladungen verantwortlich sind. Durch diese Mechanismen erfolgt eine pathologische Synchronisation der kortikalen Ströme. Synchrone Ströme können, wie bereits beschrieben, schließlich zu einem epileptischen Anfall führen.
HCN und Epilepsie beim Menschen
Obwohl Ratten- und Maus-Modelle eine pathophysiologische Rolle von HCN-Kanälen klar aufzeigen, ist ein Nachweis für eine Rolle bei menschlicher Epilepsie derzeit noch nicht erbracht. Änderungen in der Expression von HCN-Kanälen beim Menschen konnten bis dato nur im Endstadium einer Krankheit nachgewiesen werden[34] und könnten daher auch auf regulative Mechanismen zurückzuführen sein. Die Erforschung der Rolle von HCN in den verschiedenen Formen der Epilepsie wird durch verschiedene Umstände erschwert. So zeigen verschiedene Tiermodelle für unterschiedliche Epilepsie-Formen, dass sowohl eine Hoch- als auch eine Runterregulation von HCN-Untereinheiten mit Epilepsie assoziiert sein können. Die unterschiedlichen Untereinheiten sind dabei mit verschiedenen Anteilen an den jeweiligen Krankheiten beteiligt und zwar auch abhängig von ihrer zellulären Lokalisation. Daher muss eine Rolle von HCN bei Epilepsie stets sehr differenziert und auf die spezifische Epilepsie betrachtet werden. Die Forschung in diese Richtung wird in Zukunft weitere Erkenntnisse offenbaren.
Neuralgie
Verschiedene Beobachtungen lassen darauf schließen, dass HCN-Kanäle eine wichtige Rolle bei der Entstehung von Neuralgien spielen. So könnten in den dorsalen Wurzelganglien-Neuronen von Maus und Ratte eine Expression von HCN1, HCN2 und HCN3 nachgewiesen werden. Bei einer Verletzung dieser Neurone erhöht sich die Stromdichte.[71] [72] [73] Die daraufhin entstandenen Schmerzen konnten ursächlich mit dem HCN-Kanalblocker ZD7288 behandelt werden.[74] [75] Der Mechanismus, warum die HCN-Stromdichte bei einer Verletzung hochreguliert wird, ist derzeit noch nicht bekannt. Interessanterweise wird die Expression der Kanäle bei einer Verletzung herunter reguliert.[71] Wie dies mit der erhöhten Stromdichte vereinbar ist, bleibt zu untersuchen.
Kardiale Arrhythmie
Bisher sind 4 vererbte Mutationen an HCN-Kanälen in der wissenschaftlichen Literatur beschrieben. Interessanterweise finden sich all diese Mutationen in der HCN4-Untereinheit. Sie führen zu Sinus-Bradykardien. Bisher wurden nur heterozyogte Merkmalsträger beschrieben. Dies mag auf die schwerwiegenden Effekte einer Mutation auf beiden Allelen zurückzuführen sein, die sich unter anderem in der frühen Letalität von HCN4-Knock-Out-Mäusen zeigt. Die beschriebenen Mutationen sind G480R[76] und S672R[77], die beide zu einer Verschiebung der Aktivierungskurve zu stärker hyperpolarisierten Potentialen führen, 573X[78], bei der ein großer Teil des C-Terminus fehlt und die nicht mehr auf eine Änderung in der cAMP-Konzentration reagieren kann und D553N[79]. Diese Mutation führt zu einer stark reduzierten Oberflächenexpression des Kanals. Obwohl HCN4 auch im Zentralnervensystem exprimiert ist, zeigt sich bei diesen Patienten kein neuronaler Phänotyp.
Für die Untereinheiten HCN1-3 wird spekuliert, ob eine Mutation beim Menschen stets letal ist, im Gegensatz zur Maus, wo der Knock-Out toleriert wird. Alternativ ist es denkbar, dass Mutationen dieser Untereinheiten auf Grund ihrer Seltenheit bisher nicht entdeckt wurden.
HCN-Kanäle in der medizinischen Therapie
Kardiologie
Eine zu hohe Herzfrequenz ist positiv korreliert mit dem Auftreten von Krankheiten wie Ischämie und Bluthochdruck. Durch ihre große Rolle bei der Generation des Herzrhythmus sind HCN-Kanäle natürlich potentielle Angriffspunkte für eine ursächliche Therapie. Ein spezifischer Wirkstoff für HCN-Kanäle wäre zudem im Vergleich zur derzeitigen Therapien wie β-Blockern wesentlich verträglicher, da HCN-Kanäle aufgrund ihres Expressionsprofils spezifisch im Herzen angesprochen werden können. Bisherige Therapien litten meist unter Nebenwirkungen auf die Gefäße und die Atemwege. Eine aktuelle Entwicklung in diese Richtung ist das Medikament Ivabradin, das als erster HCN-Blocker für therapeutische Zwecke zugelassen wurde. Die Substanz blockiert im niedrigen mikromolaren Bereich (IC50 ≈1–2 μM) spezifisch HCN-Kanäle im Sinusknoten. Es setzt sich von der Innenseite in die Pore des HCN-Kanals. Dadurch kommt es zu einer verlangsamten Depolarisation der Zellen im Sinusknoten und somit zu einer niedrigeren Herzfrequenz. Es wird derzeit schon als Therapeutikum für Angina pectoris eingesetzt.[80] Neben Ivabradin existieren noch eine Reihe weiterer Blocker des HCN-Stroms, zum Beispiel Zatebradin oder ZD7288, das häufig experimentell genutzt wird. Allerdings ist derzeit nur Ivabradin zur Therapie beim Menschen zugelassen, da die anderen Substanzen entweder nicht spezifisch genug für kardiale HCN-Ströme sind, was zu unangenehmen neuronalen Nebenwirkungen führen kann[81], oder aber sie blockieren neben HCN-Kanälen andere Ionenkanäle.
Neurologie
Wie im Abschnitt über die Pathophysiologie bereits angesprochen, sind HCN-Kanäle auch an der Entstehung von Neuralgien beteiligt. Es ist daher nicht verwunderlich, dass spezifische Blocker von großem Nutzen in der analgetischen Therapie dieser Neuralgien sein können. Eine systemische Gabe eines solchen Blockers wurde aber definitiv kardiale Nebenwirkungen hervorrufen. Um diese Nebenwirkungen zu umgehen, wären Subtyp-spezifische Blocker für die neuronal exprimierten Untereinheiten HCN1und HCN2 hilfreich, die nicht auf HCN4-Kanäle wirken. Derzeit gibt es diese Substanzen nicht, allerdings ist eine derartige Entwicklung nicht unmöglich. Neben der Therapie von Neuralgien werden neuronale HCN-Agenzien auch im Kontext von Epilepsien diskutiert. Hier ist die mechanistische Seite jedoch noch nicht gut genug geklärt. Je nach Form der Epilepsie wäre entweder eine Blockade oder eine Aktivierung von HCN-Strömen hilfreich. Dies wurde für unterschiedliche Formen der Epilepsie bereits im Tiermodell demonstriert.[82] [83]
Weiterführende Literatur
- M. Biel, C. Wahl-Schott, S. Michalakis, X. Zong: Hyperpolarization-activated cation channels: from genes to function. In: Physiological reviews Band 89, Nummer 3, Juli 2009, S. 847–885, ISSN 0031-9333. doi:10.1152/physrev.00029.2008. PMID 19584315. (Review).
Einzelnachweise
- ↑ F. H. Yu, V. Yarov-Yarovoy, G. A. Gutman, W. A. Catterall: Overview of molecular relationships in the voltage-gated ion channel superfamily. In: Pharmacological reviews Band 57, Nummer 4, Dezember 2005, S. 387–395, ISSN 0031-6997. doi:10.1124/pr.57.4.13. PMID 16382097. (Review).
- ↑ a b c C. Wahl-Schott, M. Biel: HCN channels: structure, cellular regulation and physiological function. In: Cellular and molecular life sciences : CMLS Band 66, Nummer 3, Februar 2009, S. 470–494, ISSN 1420-9071. doi:10.1007/s00018-008-8525-0. PMID 18953682. (Review).
- ↑ W. N. Zagotta, N. B. Olivier, K. D. Black, E. C. Young, R. Olson, E. Gouaux: Structural basis for modulation and agonist specificity of HCN pacemaker channels. In: Nature Band 425, Nummer 6954, September 2003, S. 200–205, ISSN 1476-4687. doi:10.1038/nature01922. PMID 12968185.
- ↑ a b L. Zhou, S. A. Siegelbaum: Gating of HCN channels by cyclic nucleotides: residue contacts that underlie ligand binding, selectivity, and efficacy. In: Structure (London, England : 1993) Band 15, Nummer 6, Juni 2007, S. 655–670, ISSN 0969-2126. doi:10.1016/j.str.2007.04.012. PMID 17562313. PMC PMC1995447.
- ↑ B. J. Wainger, M. DeGennaro, B. Santoro, S. A. Siegelbaum, G. R. Tibbs: Molecular mechanism of cAMP modulation of HCN pacemaker channels. In: Nature Band 411, Nummer 6839, Juni 2001, S. 805–810, ISSN 0028-0836. doi:10.1038/35081088. PMID 11459060.
- ↑ J. Wang, S. Chen, S. A. Siegelbaum: Regulation of hyperpolarization-activated HCN channel gating and cAMP modulation due to interactions of COOH terminus and core transmembrane regions. In: The Journal of general physiology Band 118, Nummer 3, September 2001, S. 237–250, ISSN 0022-1295. PMID 11524455. PMC PMC2229504.
- ↑ K. B. Craven, W. N. Zagotta: CNG and HCN channels: two peas, one pod. In: Annual review of physiology Band 68, 2006, S. 375–401, ISSN 0066-4278. doi:10.1146/annurev.physiol.68.040104.134728. PMID 16460277. (Review).
- ↑ B. Much, C. Wahl-Schott, X. Zong, A. Schneider, L. Baumann, S. Moosmang, A. Ludwig, M. Biel: Role of subunit heteromerization and N-linked glycosylation in the formation of functional hyperpolarization-activated cyclic nucleotide-gated channels. In: The Journal of biological chemistry Band 278, Nummer 44, Oktober 2003, S. 43781–43786, ISSN 0021-9258. doi:10.1074/jbc.M306958200. PMID 12928435.
- ↑ a b J. Chen, J. S. Mitcheson, M. Lin, M. C. Sanguinetti: Functional roles of charged residues in the putative voltage sensor of the HCN2 pacemaker channel. In: The Journal of biological chemistry Band 275, Nummer 46, November 2000, S. 36465–36471, ISSN 0021-9258. doi:10.1074/jbc.M007034200. PMID 10962006.
- ↑ R. Männikkö, F. Elinder, H. P. Larsson: Voltage-sensing mechanism is conserved among ion channels gated by opposite voltages. In: Nature Band 419, Nummer 6909, Oktober 2002, S. 837–841, ISSN 0028-0836. doi:10.1038/nature01038. PMID 12397358.
- ↑ N. Decher, J. Chen, M. C. Sanguinetti: Voltage-dependent gating of hyperpolarization-activated, cyclic nucleotide-gated pacemaker channels: molecular coupling between the S4-S5 and C-linkers. In: The Journal of biological chemistry Band 279, Nummer 14, April 2004, S. 13859–13865, ISSN 0021-9258. doi:10.1074/jbc.M313704200. PMID 14726518.
- ↑ D. L. Prole, G. Yellen: Reversal of HCN channel voltage dependence via bridging of the S4-S5 linker and Post-S6. In: The Journal of general physiology Band 128, Nummer 3, September 2006, S. 273–282, ISSN 0022-1295. doi:10.1085/jgp.200609590. PMID 16908727. PMC PMC2151568.
- ↑ C. Ulens, J. Tytgat: Functional heteromerization of HCN1 and HCN2 pacemaker channels. In: The Journal of biological chemistry Band 276, Nummer 9, März 2001, S. 6069–6072, ISSN 0021-9258. doi:10.1074/jbc.C000738200. PMID 11133998.
- ↑ a b c H. Yu, J. Wu, I. Potapova, R. T. Wymore, B. Holmes, J. Zuckerman, Z. Pan, H. Wang, W. Shi, R. B. Robinson, M. R. El-Maghrabi, W. Benjamin, J. Dixon, D. McKinnon, I. S. Cohen, R. Wymore: MinK-related peptide 1: A beta subunit for the HCN ion channel subunit family enhances expression and speeds activation. In: Circulation research Band 88, Nummer 12, Juni 2001, S. E84–E87, ISSN 1524-4571. PMID 11420311.
- ↑ a b c C. Altomare, B. Terragni, C. Brioschi, R. Milanesi, C. Pagliuca, C. Viscomi, A. Moroni, M. Baruscotti, D. DiFrancesco: Heteromeric HCN1-HCN4 channels: a comparison with native pacemaker channels from the rabbit sinoatrial node. In: The Journal of physiology Band 549, Pt 2Juni 2003, S. 347–359, ISSN 0022-3751. doi:10.1113/jphysiol.2002.027698. PMID 12702747. PMC PMC2342966.
- ↑ a b c J. Stieber, G. Stöckl, S. Herrmann, B. Hassfurth, F. Hofmann: Functional expression of the human HCN3 channel. In: The Journal of biological chemistry Band 280, Nummer 41, Oktober 2005, S. 34635–34643, ISSN 0021-9258. doi:10.1074/jbc.M502508200. PMID 16043489.
- ↑ R. Männikkö, S. Pandey, H. P. Larsson, F. Elinder: Hysteresis in the voltage dependence of HCN channels: conversion between two modes affects pacemaker properties. In: The Journal of general physiology Band 125, Nummer 3, März 2005, S. 305–326, ISSN 0022-1295. doi:10.1085/jgp.200409130. PMID 15710913. PMC PMC2234019.
- ↑ F. Elinder, R. Männikkö, S. Pandey, H. P. Larsson: Mode shifts in the voltage gating of the mouse and human HCN2 and HCN4 channels. In: The Journal of physiology Band 575, Pt 2September 2006, S. 417–431, ISSN 0022-3751. doi:10.1113/jphysiol.2006.110437. PMID 16777944. PMC PMC1819464.
- ↑ A. Bruening-Wright, H. P. Larsson: Slow conformational changes of the voltage sensor during the mode shift in hyperpolarization-activated cyclic-nucleotide-gated channels. In: The Journal of neuroscience : the official journal of the Society for Neuroscience Band 27, Nummer 2, Januar 2007, S. 270–278, ISSN 1529-2401. doi:10.1523/JNEUROSCI.3801-06.2007. PMID 17215386.
- ↑ a b B. Santoro, D. T. Liu, H. Yao, D. Bartsch, E. R. Kandel, S. A. Siegelbaum, G. R. Tibbs: Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. In: Cell Band 93, Nummer 5, Mai 1998, S. 717–729, ISSN 0092-8674. PMID 9630217.
- ↑ a b c d A. Ludwig, X. Zong, J. Stieber, R. Hullin, F. Hofmann, M. Biel: Two pacemaker channels from human heart with profoundly different activation kinetics. In: The EMBO journal Band 18, Nummer 9, Mai 1999, S. 2323–2329, ISSN 0261-4189. doi:10.1093/emboj/18.9.2323. PMID 10228147. PMC PMC1171315.
- ↑ B. Santoro, S. Chen, A. Luthi, P. Pavlidis, G. P. Shumyatsky, G. R. Tibbs, S. A. Siegelbaum: Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. In: The Journal of neuroscience : the official journal of the Society for Neuroscience Band 20, Nummer 14, Juli 2000, S. 5264–5275, ISSN 0270-6474. PMID 10884310.
- ↑ D. DiFrancesco: Characterization of single pacemaker channels in cardiac sino-atrial node cells. In: Nature Band 324, Nummer 6096, 1986, S. 470–473, ISSN 0028-0836. doi:10.1038/324470a0. PMID 2431323.
- ↑ a b G. Michels, F. Er, I. Khan, M. Südkamp, S. Herzig, U. C. Hoppe: Single-channel properties support a potential contribution of hyperpolarization-activated cyclic nucleotide-gated channels and If to cardiac arrhythmias. In: Circulation Band 111, Nummer 4, Februar 2005, S. 399–404, ISSN 1524-4539. doi:10.1161/01.CIR.0000153799.65783.3A. PMID 15687126.
- ↑ H. C. Pape: Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. In: Annual review of physiology Band 58, 1996, S. 299–327, ISSN 0066-4278. doi:10.1146/annurev.ph.58.030196.001503. PMID 8815797. (Review).
- ↑ X. Yu, K. L. Duan, C. F. Shang, H. G. Yu, Z. Zhou: Calcium influx through hyperpolarization-activated cation channels (I(h) channels) contributes to activity-evoked neuronal secretion. In: Proceedings of the National Academy of Sciences of the United States of America Band 101, Nummer 4, Januar 2004, S. 1051–1056, ISSN 0027-8424. doi:10.1073/pnas.0305167101. PMID 14724293. PMC PMC327149.
- ↑ X. Yu, X. W. Chen, P. Zhou, L. Yao, T. Liu, B. Zhang, Y. Li, H. Zheng, L. H. Zheng, C. X. Zhang, I. Bruce, J. B. Ge, S. Q. Wang, Z. A. Hu, H. G. Yu, Z. Zhou: Calcium influx through If channels in rat ventricular myocytes. In: American journal of physiology. Cell physiology Band 292, Nummer 3, März 2007, S. C1147–C1155, ISSN 0363-6143. doi:10.1152/ajpcell.00598.2005. PMID 17065201. PMC PMC1849975.
- ↑ A. M. Frace, F. Maruoka, A. Noma: External K+ increases Na+ conductance of the hyperpolarization-activated current in rabbit cardiac pacemaker cells. In: Pflügers Archiv : European journal of physiology Band 421, Nummer 2–3, Juni 1992, S. 97–99, ISSN 0031-6768. PMID 1326752.
- ↑ L. P. Wollmuth, B. Hille: Ionic selectivity of Ih channels of rod photoreceptors in tiger salamanders. In: The Journal of general physiology Band 100, Nummer 5, November 1992, S. 749–765, ISSN 0022-1295. PMID 1282144. PMC PMC2229118.
- ↑ D. DiFrancesco: A study of the ionic nature of the pace-maker current in calf Purkinje fibres. In: The Journal of physiology Band 314, Mai 1981, S. 377–393, ISSN 0022-3751. PMID 6273534. PMC PMC1249440.
- ↑ a b A. M. Frace, F. Maruoka, A. Noma: Control of the hyperpolarization-activated cation current by external anions in rabbit sino-atrial node cells. In: The Journal of physiology Band 453, 1992, S. 307–318, ISSN 0022-3751. PMID 1281504. PMC PMC1175559.
- ↑ a b C. Viscomi, C. Altomare, A. Bucchi, E. Camatini, M. Baruscotti, A. Moroni, D. DiFrancesco: C terminus-mediated control of voltage and cAMP gating of hyperpolarization-activated cyclic nucleotide-gated channels. In: The Journal of biological chemistry Band 276, Nummer 32, August 2001, S. 29930–29934, ISSN 0021-9258. doi:10.1074/jbc.M103971200. PMID 11397812.
- ↑ A. Ludwig, X. Zong, M. Jeglitsch, F. Hofmann, M. Biel: A family of hyperpolarization-activated mammalian cation channels. In: Nature Band 393, Nummer 6685, Juni 1998, S. 587–591, ISSN 0028-0836. doi:10.1038/31255. PMID 9634236.
- ↑ a b c d e S. Moosmang, M. Biel, F. Hofmann, A. Ludwig: Differential distribution of four hyperpolarization-activated cation channels in mouse brain. In: Biological chemistry Band 380, Nummer 7–8, 1999 Jul-Aug, S. 975–980, ISSN 1431-6730. doi:10.1515/BC.1999.121. PMID 10494850.
- ↑ a b c W. Shi, R. Wymore, H. Yu, J. Wu, R. T. Wymore, Z. Pan, R. B. Robinson, J. E. Dixon, D. McKinnon, I. S. Cohen: Distribution and prevalence of hyperpolarization-activated cation channel (HCN) mRNA expression in cardiac tissues. In: Circulation research Band 85, Nummer 1, Juli 1999, S. e1–e6, ISSN 1524-4571. PMID 10400919.
- ↑ a b N. J. Chandler, I. D. Greener, J. O. Tellez, S. Inada, H. Musa, P. Molenaar, D. Difrancesco, M. Baruscotti, R. Longhi, R. H. Anderson, R. Billeter, V. Sharma, D. C. Sigg, M. R. Boyett, H. Dobrzynski: Molecular architecture of the human sinus node: insights into the function of the cardiac pacemaker. In: Circulation Band 119, Nummer 12, März 2009, S. 1562–1575, ISSN 1524-4539. doi:10.1161/CIRCULATIONAHA.108.804369. PMID 19289639.
- ↑ T. Notomi, R. Shigemoto: Immunohistochemical localization of Ih channel subunits, HCN1-4, in the rat brain. In: The Journal of comparative neurology Band 471, Nummer 3, April 2004, S. 241–276, ISSN 0021-9967. doi:10.1002/cne.11039. PMID 14991560.
- ↑ a b c A. Ludwig, T. Budde, J. Stieber, S. Moosmang, C. Wahl, K. Holthoff, A. Langebartels, C. Wotjak, T. Munsch, X. Zong, S. Feil, R. Feil, M. Lancel, K. R. Chien, A. Konnerth, H. C. Pape, M. Biel, F. Hofmann: Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. In: The EMBO journal Band 22, Nummer 2, Januar 2003, S. 216–224, ISSN 0261-4189. doi:10.1093/emboj/cdg032. PMID 12514127. PMC PMC140107.
- ↑ Wahl-Schott C, Baumann L, Zong X, Biel M. An arginine residue in the pore region is a key determinant of chloride dependence in cardiac pacemaker channels. J Biol Chem 280: 13694–13700, 2005
- ↑ Zong X, Stieber J, Ludwig A, Hofmann F, Biel M. A single histidine residue determines the pH sensitivity of the pacemaker channel HCN2. J Biol Chem 276: 6313–6319, 2001
- ↑ a b Stevens DR, Seifert R, Bufe B, Muller F, Kremmer E, Gauss R, Meyerhof W, Kaupp UB, Lindemann B.: Hyperpolarization-activated channels HCN1 and HCN4 mediate responses to sour stimuli. Nature 413: 631–635, 2001
- ↑ Qu J, Barbuti A, Protas L, Santoro B, Cohen IS, Robinson RB. HCN2 overexpression in newborn and adult ventricular myocytes: distinct effects on gating and excitability. Circ Res 89: E8–14, 2001
- ↑ Robinson RB, Yu H, Chang F, Cohen IS. Developmental change in the voltage-dependence of the pacemaker current, if, in rat ventricle cells. Pflügers Arch 433: 533–535, 1997
- ↑ Wu JY, Cohen IS. Tyrosine kinase inhibition reduces i(f) in rabbit sinoatrial node myocytes. Pflügers Arch 434: 509–514, 1997
- ↑ Poolos NP, Bullis JB, Roth MK. Modulation of h-channels in hippocampal pyramidal neurons by p38 mitogenactivated protein kinase. J Neurosci 26: 7995–8003, 2006
- ↑ Santoro B, Grant SG, Bartsch D, Kandel ER. Interactive cloning with the SH3 domain of N-src identifies a new brain specific ion channel protein, with homology to eag and cyclic nucleotide-gated channels. Proc Natl Acad Sci USA 94: 14815–14820, 1997
- ↑ a b Zong X, Eckert C, Yuan H, Wahl-Schott C, Abicht H, Fang L, Li R, Mistrik P, Gerstner A, Much B, Baumann L, Michalakis S, Zeng R, Chen Z, Biel M. A novel mechanism of modulation of hyperpolarization-activated cyclic nucleotide-gated channels by Src kinase. J Biol Chem 280: 34224–34232, 2005
- ↑ Arinsburg SS, Cohen IS, Yu HG. Constitutively active Src tyrosine kinase changes gating of HCN4 channels through direct binding to the channel proteins. J Cardiovasc Pharmacol 47: 578–586, 2006
- ↑ Yu HG, Lu Z, Pan Z, Cohen IS. Tyrosine kinase inhibition differentially regulates heterologously expressed HCN channels. Pflügers Arch 447: 392–400, 2004
- ↑ Decher N, Bundis F, Vajna R, Steinmeyer K. KCNE2 modulates current amplitudes and activation kinetics of HCN4: influence of KCNE family members on HCN4 currents. Pflügers Arch 446: 633–640, 2003
- ↑ Qu J, Kryukova Y, Potapova IA, Doronin SV, Larsen M, Krishnamurthy G, Cohen IS, Robinson RB. MiRP1 modulates HCN2 channel expression and gating in cardiac myocytes. J Biol Chem 279: 43497–43502, 2004
- ↑ Michels G, Er F, Khan IF, Endres-Becker J, Brandt MC, Gassanov N, Johns DC, Hoppe UC. K channel regulator KCR1 suppresses heart rhythm by modulating the pacemaker current I(f). PLoS ONE 3: e1511, 2008
- ↑ Williams SR, Christensen SR, Stuart GJ, Hausser M. Membrane potential bistability is controlled by the hyperpolarizationactivated current I(H) in rat cerebellar Purkinje neurons in vitro. J Physiol 539: 469–83, 2002
- ↑ Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci 18: 7613–7624, 1998
- ↑ Magee JC. Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci 2: 848, 1999
- ↑ Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, Vijayraghavan S, Brennan A, Dudley A, Nou E, Mazer JA, McCormick DA, Arnsten AF. Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell 129: 397–410, 2007
- ↑ Nolan MF, Malleret G, Lee KH, Gibbs E, Dudman JT, Santoro B, Yin D, Thompson RF, Siegelbaum SA, Kandel ER, Morozov A. The hyperpolarization-activated HCN1 channel is important for motor learning and neuronal integration by cerebellar Purkinje cells. Cell 115: 551–564, 2003
- ↑ DiFrancesco D, Tortora P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature 351: 145–147, 1991
- ↑ M. F. Nolan, G. Malleret, J. T. Dudman, D. L. Buhl, B. Santoro, E. Gibbs, S. Vronskaya, G. Buzsáki, S. A. Siegelbaum, E. R. Kandel, A. Morozov: A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. In: Cell Band 119, Nummer 5, November 2004, S. 719–732, ISSN 0092-8674. doi:10.1016/j.cell.2004.11.020. PMID 15550252.
- ↑ a b S. Herrmann, J. Stieber, A. Ludwig: Pathophysiology of HCN channels. In: Pflügers Archiv: European journal of physiology Band 454, Nummer 4, Juli 2007, S. 517–522, ISSN 0031-6768. doi:10.1007/s00424-007-0224-4. PMID 17549513. (Review).
- ↑ J. Stieber, S. Herrmann, S. Feil, J. Löster, R. Feil, M. Biel, F. Hofmann, A. Ludwig: The hyperpolarization-activated channel HCN4 is required for the generation of pacemaker action potentials in the embryonic heart. In: Proceedings of the National Academy of Sciences of the United States of America Band 100, Nummer 25, Dezember 2003, S. 15235–15240, ISSN 0027-8424. doi:10.1073/pnas.2434235100. PMID 14657344. PMC PMC299971.
- ↑ J. Stieber, K. Wieland, G. Stöckl, A. Ludwig, F. Hofmann: Bradycardic and proarrhythmic properties of sinus node inhibitors. In: Molecular pharmacology Band 69, Nummer 4, April 2006, S. 1328–1337, ISSN 0026-895X. doi:10.1124/mol.105.020701. PMID 16387796.
- ↑ E. L. van Luijtelaar, A. M. Coenen: Two types of electrocortical paroxysms in an inbred strain of rats. In: Neuroscience letters Band 70, Nummer 3, Oktober 1986, S. 393–397, ISSN 0304-3940. PMID 3095713.
- ↑ W. H. Drinkenburg, E. L. van Luijtelaar, W. J. van Schaijk, A. M. Coenen: Aberrant transients in the EEG of epileptic rats: a spectral analytical approach. In: Physiology & behavior Band 54, Nummer 4, Oktober 1993, S. 779–783, ISSN 0031-9384. PMID 8248357.
- ↑ D. Gauguier, G. van Luijtelaar, M. T. Bihoreau, S. P. Wilder, R. F. Godfrey, J. Vossen, A. Coenen, R. D. Cox: Chromosomal mapping of genetic loci controlling absence epilepsy phenotypes in the WAG/Rij rat. In: Epilepsia Band 45, Nummer 8, August 2004, S. 908–915, ISSN 0013-9580. doi:10.1111/j.0013-9580.2004.13104.x. PMID 15270755.
- ↑ S. Hirose, A. Mitsudome, M. Okada, S. Kaneko: Genetics of idiopathic epilepsies. In: Epilepsia Band 46 Suppl 1, 2005, S. 38–43, ISSN 0013-9580. doi:10.1111/j.0013-9580.2005.461011.x. PMID 15816978. (Review).
- ↑ G. Gandolfo, S. Romettino, C. Gottesmann, G. van Luijtelaar, A. Coenen, J. N. Bidard, M. Lazdunski: K+ channel openers decrease seizures in genetically epileptic rats. In: European journal of pharmacology Band 167, Nummer 1, August 1989, S. 181–183, ISSN 0014-2999. PMID 2506066.
- ↑ E. Tsakiridou, L. Bertollini, M. de Curtis, G. Avanzini, H. C. Pape: Selective increase in T-type calcium conductance of reticular thalamic neurons in a rat model of absence epilepsy. In: The Journal of neuroscience : the official journal of the Society for Neuroscience Band 15, Nummer 4, April 1995, S. 3110–3117, ISSN 0270-6474. PMID 7722649.
- ↑ T. Budde, L. Caputi, T. Kanyshkova, R. Staak, C. Abrahamczik, T. Munsch, H. C. Pape: Impaired regulation of thalamic pacemaker channels through an imbalance of subunit expression in absence epilepsy. In: The Journal of neuroscience : the official journal of the Society for Neuroscience Band 25, Nummer 43, Oktober 2005, S. 9871–9882, ISSN 1529-2401. doi:10.1523/JNEUROSCI.2590-05.2005. PMID 16251434.
- ↑ M. H. Kole, A. U. Bräuer, G. J. Stuart: Inherited cortical HCN1 channel loss amplifies dendritic calcium electrogenesis and burst firing in a rat absence epilepsy model. In: The Journal of physiology Band 578, Pt 2Januar 2007, S. 507–525, ISSN 0022-3751. doi:10.1113/jphysiol.2006.122028. PMID 17095562. PMC PMC2075144.
- ↑ a b Chaplan SR, Guo HQ, Lee DH, Luo L, Liu C, Kuei C, Velumian AA, Butler MP, Brown SM, Dubin AE. Neuronal hyperpolarization-activated pacemaker channels drive neuropathic pain. J Neurosci 23: 1169–1178, 2003
- ↑ Kitagawa J, Takeda M, Suzuki I, Kadoi J, Tsuboi Y, Honda K, Matsumoto S, Nakagawa H, Tanabe A, Iwata K. Mechanisms involved in modulation of trigeminal primary afferent activity in rats with peripheral mononeuropathy. Eur J Neurosci 24: 1976–1986, 2006
- ↑ Yao H, Donnelly DF, Ma C, LaMotte RH. Upregulation of the hyperpolarization-activated cation current after chronic compression of the dorsal root ganglion. J Neurosci 23: 2069–2074, 2003
- ↑ Lee DH, Chang L, Sorkin LS, Chaplan SR. Hyperpolarization-activated, cation-nonselective, cyclic nucleotidemodulated channel blockade alleviates mechanical allodynia and suppresses ectopic discharge in spinal nerve ligated rats. J Pain 6: 417–424, 2005
- ↑ Sun Q, Xing GG, Tu HY, Han JS, Wan Y. Inhibition of hyperpolarization-activated current by ZD7288 suppresses ectopic discharges of injured dorsal root ganglion neurons in a rat model of neuropathic pain. Brain Res 1032: 63–69, 2005
- ↑ Nof E, Luria D, Brass D, Marek D, Lahat H, Reznik-Wolf H, Pras E, Dascal N, Eldar M, Glikson M. Point mutation in the HCN4 cardiac ion channel pore affecting synthesis, trafficking, and functional expression is associated with familial asymptomatic sinus bradycardia. Circulation 116: 463–470, 2007
- ↑ Milanesi R, Baruscotti M, Gnecchi-Ruscone T, DiFrancesco D. Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N Engl J Med 354: 151–157, 2006
- ↑ Schulze-Bahr E, Neu A, Friederich P, Kaupp UB, Breithardt G, Pongs O, Isbrandt D. Pacemaker channel dysfunction in a patient with sinus node disease. J Clin Invest 111: 1537–1545, 2003
- ↑ Ueda K, Nakamura K, Hayashi T, Inagaki N, Takahashi M, Arimura T, Morita H, Higashiuesato Y, Hirano Y, Yasunami M, Takishita S, Yamashina A, Ohe T, Sunamori M, Hiraoka M, Kimura A. Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J Biol Chem 279: 27194–27198, 2004
- ↑ Sulfi S, Timmis AD. Ivabradine: the first selective sinus node I(f) channel inhibitor in the treatment of stable angina. Int J Clin Pract 60: 222–228, 2006
- ↑ Cervetto L, Demontis GC, Gargini C. Cellular mechanisms underlying the pharmacological induction of phosphenes. Br J Pharmacol 150: 383–390, 2007
- ↑ Kitayama M, Miyata H, Yano M, Saito N, Matsuda Y, Yamauchi T, Kogure S. Ih blockers have a potential of antiepileptic effects. Epilepsia 44: 20–24, 2003
- ↑ Surges R, Freiman TM, Feuerstein TJ. Gabapentin increases the hyperpolarization-activated cation current Ih in rat CA1 pyramidal cells. Epilepsia 44: 150–156, 2003
Wikimedia Foundation.