- Heterogene Katalyse
-
Heterogene Katalyse ist eine Form der Katalyse, bei der der Katalysator und die reagierenden Stoffe einer chemischen Reaktion in unterschiedlichen Phasen vorliegen.
Die Entwicklung und Umsetzung heterogen-katalytischer Verfahren zur Herstellung von Grundchemikalien, etwa dem Kontaktverfahren zur Schwefelsäureherstellung, dem Haber-Bosch-Verfahren zur Ammoniaksynthese oder der Methanolherstellung haben einen entscheidenden Anteil am geschäftlichen Erfolg der chemischen Industrie.
In Erdölraffinerien liefern heterogen-katalytische Verfahren einen erheblichen Beitrag zur Gewinnung hochoktaniger Ottokraftstoffe und anderer wertvoller Kohlenwasserstoffkomponenten. Im Umweltbereich dient die heterogene Katalyse der Reinhaltung der Luft durch die Reduktion und Entfernung von Schadstoffen aus Kraftwerks- und Automobilemissionen.
Geschichte der Heterogenen Katalyse
 Porträt Joseph Priestleys, gemalt von Rembrandt Peale, 1801
Porträt Joseph Priestleys, gemalt von Rembrandt Peale, 1801
 Fritz Haber
Fritz HaberAls eines der ersten Beispiele der heterogenen Katalyse gilt die im Jahr 1783 gemachte Beobachtung Joseph Priestleys, der den Zerfall von Ethanol zu Ethylen und Wasser an Tonerde beschrieb. [1] Martinus van Marum beschrieb 1796 die katalytische Dehydrierung von Alkohol zu Aldehyden an glühenden Metallen wie Kupfer. [1] Nach weiteren Entdeckungen wie den Ammoniakzerfall zu Stickstoff und Wasserstoff an Eisenkatalysatoren durch Berthollet und den Zerfall von Wasserstoffperoxid an Silber, Silberoxid und Mangandioxid durch Thénard führte die 1823 gefundene Entzündung von Wasserstoff an Platin durch Johann Wolfgang Döbereiner zur Entwicklung des Döbereiners Feuerzeug. Diese wurde in relativ großen Stückzahlen hergestellt und fand bis Mitte des 18. Jahrhunderts Verwendung.[2]
Im Jahr 1835 erkannte Jöns Jakob Berzelius in diesen Reaktionen die Gemeinsamkeit, dass neben den Edukten und Produkten immer ein weiterer Stoff in der Reaktion notwendig war, der offenbar nicht verbraucht wurde. Er prägte dazu den Begriff Katalyse '.[3]
Im Jahr 1894 definierte Wilhelm Ostwald den Vorgang der Katalyse wie folgt:[4]
„Katalyse ist die Beschleunigung eines langsam verlaufenden chemischen Vorgangs durch die Gegenwart eines fremden Stoffes“
und
„Ein Katalysator ist ein Stoff, der die Geschwindigkeit einer chemischen Reaktion erhöht, ohne selbst dabei verbraucht zu werden und ohne die endgültige Lage des thermodynamischen Gleichgewichts dieser Reaktion zu verändern.“
– Wilhelm Ostwald
Als Anerkennung für seine Arbeiten über die Katalyse wurde Ostwald im Jahre 1909 mit dem Nobelpreis für Chemie ausgezeichnet.
In der chemischen Industrie fand die die heterogene Katalyse in der Schwefelsäureherstellung nach dem Kontaktverfahren die erste großtechnische Anwendung. [5] Bereits 1901 wurde die Fetthärtung durch katalytische Hydrierung von Ölsäure zu Stearinsäure mit Wasserstoff an fein verteiltem Nickel durch Wilhelm Normann entwickelt.[6] Sie stellt die Grundlage der großindustriellen Margarineherstellung dar, das bereits 1909 im großtechnischen Einsatz war.
Im frühen 20. Jahrhundert begann die Entwicklung einer Reihe von Verfahren, die bis heute zu den wichtigsten der chemischen Industrie zählen. Fritz Haber, Carl Bosch und Alwin Mittasch entwickelten 1910 die Ammoniak-Synthese aus den Elementen Stickstoff und Wasserstoff an heterogenen Eisen-Kontakten, das Haber-Bosch-Verfahren. Wilhelm Ostwald entwickelte das Ostwald-Verfahren der Ammoniak-Oxidation an Platin-Netzen zu Salpetersäure, wodurch der zuvor knappe Nitrat-Dünger im großen Maßstab zur Verfügung stand. Ebenfalls bei der BASF im Jahr 1923 entwickelte Matthias Pier ein katalytisches Hochdruckverfahren zur Synthese von Methanol aus Synthesegas an heterogenen Zinkoxid-Chromoxid-Katalysatoren.[7]
Auf dem Gebiet der Raffinierietechnologie wurden weitere heterogenkatalytische Verfahren entwickelt. Durch Katalytisches Reforming von niedrigoktanigen Alkanen an heterogenen Platin-Zinn- oder Platin-Rhenium auf Aluminiumoxid-Kontakten entstanden hochoktanige, aromaten- und isoalkan-reiche Benzine. Das Verfahren stellt bis heute pro Tag mehrere Millionen Liter hochoktaniges Benzin zur Verfügung. [8]
In den 1920 und 1930 Jahren führten die Studien zur Adsorption von Gasen von Irving Langmuir (Nobelpreis 1932) und später die kinetischen Studien von Cyril Norman Hinshelwood (Nobelpreis 1956) zum besseren Verständnis der Mechanismen von heterogen katalysierten Reaktionen von zwei co-adsorbierten Reaktanden (Langmuir-Hinshelwood-Mechanismus). [9] Im Jahre 1943 entwickelten Eley und Rideal einen Mechanismus für Reaktionen, bei denen einer der Reaktanden adsorbiert ist und der zweite aus der Gasphase reagiert.[10]
In der Umwelttechnik wurde 1957 für die Entstickung von Kraftwerksemissionen das Verfahren der selektiven katalytische Reduktion (SCR) von Stickoxiden mit Ammoniak als Reduktionsmittel von der Engelhard Corporation entwickelt.[11] Ein entscheidender Beitrag zur Reinhaltung der Luft war 1973 die Entwicklung des Drei-Wege-Katalysators durch Carl D. Keith und John J. Mooney.[12]
 Gerhard Ertl
Gerhard ErtlWissenschaftler wie Gerhard Ertl und Gábor A. Somorjai untersuchten seit den 1960er Jahren die Elementarschritte von Oberflächenreaktionen und die Struktur von Adsorbaten an Modellkatalysatoren wie Einkristalloberflächen unter Anwendung moderner oberflächenanalytischer Methoden. So wurde der Mechanismus der Ammoniaksynthese durch Ertl in den 1970er-Jahren aufgeklärt.
In den 1970er Jahre wurde ein Reihe neuartige Zeolithe entwickelt mit bis dahin völlig unbekannten Eigenschaften. Im Jahr 1972 gelang es Mitarbeitern der Firma Mobil Oil die Grundlage für eine ganze Reihe neuer, als Pentasile bezeichnete Zeolithe zu schaffen. Wichtigster Vertreter der Pentasile ist der „ZSM-5“, der in petrochemischen Prozessen wie Methanol to Gasoline Verwendung findet.[13]
Feststoffkatalysatoren
Feststoffkatalysatoren können grob in Pulver-, Formkörper- und Monolithkatalysatoren unterschieden werden.
Formkörperkatalysatoren
Die Formkörperkatalysatoren bestehen zumeist aus mit aktiver Komponente beschichteten keramischen Teilchen. Typische Dimensionen sind hier z. B. 3×3 mm bis 6×6 mm Zylinder oder Kugeln mit Durchmessern zwischen 2 bis 6 mm. [14] Die keramischen Materialien sind häufig Aluminiumoxid, Zeolithe und Siliziumdioxid. Eingesetzt werden diese Katalysatoren zumeist in sogenannten Festbettreaktoren, in denen die Edukte kontinuierlich zu- und die entstehenden Produkte abgeführt werden. Wichtige Kenngrößen sind hier neben der katalytischen Wirksamkeit die Schüttdichte und Druckverlusteigenschaften des Materials.[14]
Monolithkatalysatoren
 Monolithwaben im Größenvergleich zu einer 1 Euro Münze
Monolithwaben im Größenvergleich zu einer 1 Euro MünzeIm Falle der Monolithkatalysatoren wird ein Wabenkörper mit einem so genannten Washcoat beschichtet. Der Grundkörper besteht hierbei zumeist aus mineralischer Keramik wie Cordierit oder aus Metall. Der Washcoat ist eine Pulversuspension, zum Beispiel ein Gemisch aus Aluminiumoxid, Siliziumdioxid und andere Metalloxide. Diese, zumeist wässrige, Pulversuspension wird auf die Wabe aufgebracht, angetrocknet und danach mit einer aktiven Metallkomponente wie zum Beispiel Platin / Rhodium / Palladium bei PKW-Katalysatoren imprägniert und anschließend durch Calcinierung aktiviert. Das so genannte Washcoaten geschieht, weil der reine Träger oft nicht genügend Aufnahmekapazität für das aktive Metall aufweist. Wichtige Kenngrößen sind hier neben der katalytischen Wirksamkeit die Zelldichte also Anzahl der Kanäle pro Anströmflächeneinheit und Druckverlust des Monolithen.[15]
Pulverkatalysatoren
Pulverkatalysatoren hingegen werden zumeist in nicht- oder nur semikontinuierlichen Rührkesselreaktoren oder Wirbelschichtreaktoren eingesetzt. Das Katalysatorpulver wird durch Adsorption oder Tränken eines Trägers mit zum Beispiel Aktivkohle, Aluminiumoxid, Siliziumdioxid und andere mit einer Metallsalzlösung, die die aktive Komponente beinhaltet, hergestellt. Wichtige Kenngrößen sind hier, neben der katalytischen Wirksamkeit, die Filtrierbarkeit, die Abriebfestigkeit und die Dichte des Materials.[16]
Herstellung heterogener Katalysatoren
Der am häufigsten eingesetzte Aggregatzustand heterogener Katalysatoren ist die feste Form. Hierbei besteht der Katalysator oder Kontakt entweder vollständig aus der aktiven Komponente, sogenannte Vollkatalysatoren, oder die wirksame aktive Komponente wird auf ein Trägermaterial aufgebracht. Die Verfahrensparameter der durchgeführten chemischen Reaktion stellen eine Reihe von Anforderungen an die physikalischen und chemischen Eigenschaften der Kontakte. Die Katalysatorentwicklung muss daher eine zweckmäßige Herstellungsmethode entwickeln, welche die gewünschten chemischen und physikalischen Eigenschaften der Kontakte liefert.
Im Zusammenspiel mit den reaktionstechnischen Bedingungen muss versucht werden, die Aktivität, Selektivität und Raum-Zeit-Ausbeute des Kontakts zu optimieren. Neben den chemisch wichtigen Eigenschaften muss der Kontakt den prozesstechnischen Anforderungen entsprechen, das heißt, Eigenschaften wie die Regenerierbarkeit, die Abriebfestigkeit, die Reproduzierbarkeit der Herstellung müssen einstellbar und zu so geringen Kosten wie möglich erfolgen.
Fällung
Die Fällung (auch Präzipitation) oder Co-Fällung ist ein gängiges Verfahren zur Herstellung von Vollkatalysatoren. Ein bekanntes Beispiel ist die Herstellung von Kupfer-Zinkoxid-Alumina-Katalysatoren für die Methanolherstellung. Nach der Fällung folgen in der Regel die Schritte Waschen, Trocknen, Calcinieren und Aktivieren des Katalysators. Die Aktivität und Selektivität des fertigen Katalysators lässt sich über die chemischen und physikalischen Parameter der Herstellungsschritte beeinflussen.
Schon die Wahl der Metallsalze kann einen Einfluss auf die späteren Eigenschaften des Kontakts haben. Für die Herstellung werden in der industriellen Praxis oft die gängigen anorganischen Metallsalze wie Hydroxide, Nitrate oder Carbonate verwendet. Die Fällung kann durch verschiedene Methoden wie pH-Wert Einstellung, Übersättigung der Lösung oder der Zusatz spezieller Fällmittel ausgelöst werden.
Durch das Waschen werden Fremdbestandteile entfernt. Die Trocknung erfolgt oft im Gasstrom bei Temperaturen, die eine Zersetzung der Metallsalze vermeiden. Die Calcinierung wird bei Temperaturen zwischen 300 bis 800 °C durchgeführt und überführt in der Regel die eingesetzten Metallsalze in das Oxid. Außerdem werden gegebenenfalls für die Fällung benötigte organische Additive wie zum Beispiel Citronensäure zur Pufferung entfernt. Die Aktivierung, zum Beispiel durch Reduktion mit Wasserstoff, erfolgt oft unter Reaktionsbedingungen in situ.
Neben der Einstellung der gewünschten chemischen Zusammensetzung wird gegebenenfalls versucht, durch die Herstellungsbedingungen zum Beispiel Gitterfehler zu erzeugen, die eine höhere katalytische Aktivität aufweisen. Dies kann etwa durch die Einfügung von Heteroatomen in die aktive Phase geschehen.
Imprägnierung
 Aktivkomponentenprofile heterogener Katalysatoren
Aktivkomponentenprofile heterogener KatalysatorenImprägnierung ist eine wichtige Methode zur Herstellung industrieller Trägerkatalysatoren. Bei der Imprägnierung werden poröse Trägermaterialien mit großer innerer Oberfläche mit einer in der Regel wässrigen Lösung eines Metallsalzes versetzt. Zwei weithin bekannte Imprägnierungstechniken sind die diffusionskontrolliert Imprägnierung und die trockene Imprägnierung. In nachfolgenden Schritten wird der imprägnierte Katalysator getrocknet, und abschließend calziniert, wobei die Aktivkomponente in das Metall oder das Metalloxid überführt werden kann.
Durch geeignete Wahl der Imprägnierungsbedingungen können Profile der Aktivkomponente ausgebildet werden, die vorteilhaft für die Durchführung der chemischen Reaktion sein können. Verbreitet sind Katalysatoren mit einem einheitlichen Aktivkomponentenprofil, Schalenkatalysatoren, bei denen die Aktivkomponente nur im äußeren Bereich aufgebracht wurde oder Katalysatoren, bei denen die Aktivkomponente nur im Inneren des Katalysators liegt. Schalenkatalysatoren finden Einsatz bei Reaktionen, bei denen die Porendiffusion ein limitierender Faktor ist. Unterliegt der Katalysator während des Prozesses mechanischer Abrasion, ist es vorteilhaft, wenn teures Edelmetall nur im Inneren des Kontakts vorliegt.
Diffusionskontrollierte Imprägnierung
Im ersten Schritt wird bei der diffusionskontrollierten Imprägnierung der Kontakt mit reinem Lösungsmittels im Überschuss benetzt. Danach wird der meist wässrigen Lösung ein Metallsalz zugegeben. Die Metallionen wandern dann durch Diffusion in die Poren des Kontaktes und werden dort adsorbiert. Der Prozess der diffusionskontrollierten Imprägnierung führt häufig zu einer einheitlichen Verteilung der aktiven Komponente über das Katalysatorkorn. Im Gegensatz zur trockenen Imprägnierung ist der Zeitaufwand jedoch größer.
Trockene Imprägnierung
Bei der trockenen Imprägnierung (engl. Incipient Wetness Impregnation) oder Kapillarimprägnierung wird der Kontakt mit einem Lösungsmittel versetzt, das bereits die gelöste Aktivkomponente enthält. Die Lösungsmittelmenge ist dabei gleich oder kleiner als das Gesamtporenvolumen der Kontaktmasse, der Kontakt erscheint daher nach der Imprägnierung trocken. Die treibende Kraft der Imprägnierung ist in diesem Fall die Kapillarkraft. Durch die auftretenden Kapillarkräfte und die entstehende Adsorptionswärme können im Kontakt eingeschlossene Gase zum Teil nicht entweichen, was zu einem Zerspringen des Katalysatorkorns bei der Imprägnierung führen kann. Um dies zu verhindern, wird der Kontakt zum Teil vor der Imprägnierung unter Vakuum evakuiert.
Sol-Gel-Prozess
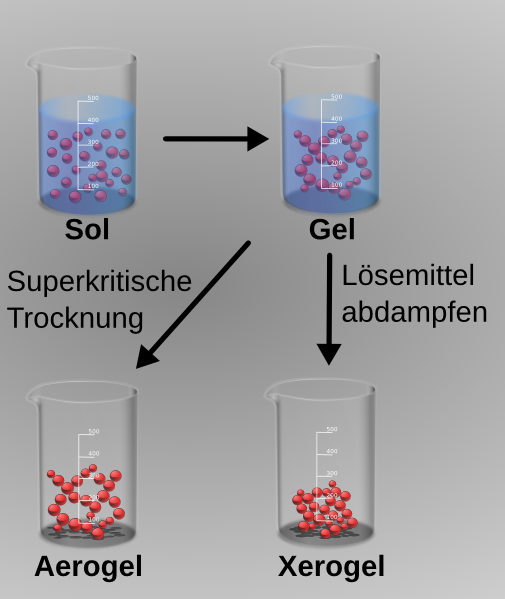
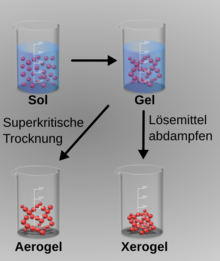 Sol, Gel, Xerogel und Aerogel
Sol, Gel, Xerogel und AerogelDer Sol-Gel-Prozess kann zur Herstellung anorganischer Katalysatoren aus kolloidalen Dispersionen, den sogenannten Solen, verwendet werden. Als Ausgangsmaterialien oder Precursor werden bevorzugt metallorganische Verbindungen wie Aluminium-(2-propylat), Aluminium-(2-butylat), Zirkonpropylat, Titanethylat oder Titan-(2-propylat) verwendet, die zunächst hydrolysiert werden. Durch Vernetzung und Trocknung lassen sich Pulver, Fasern, Schichten oder Aerogele hoher Reinheit und mit definierter Porengrößenverteilung erzeugen.
Chemische Gasphasenabscheidung
Neben dem Aufbringen der Aktivkomponente aus der flüssigen Phase kann dies auch durch die Adsorption von flüchtigen anorganischen oder metallorganischen Verbindungen aus der Gasphase erfolgen. Nach der Adsorption erfolgt die chemische oder thermische Zersetzung des Precursors unter Freisetzung der Liganden und der Fixierung des Metalls am Adsorptionsort.
Der Prozess wird meist bei höheren Temperaturen im Vakuum durchgeführt. Neben den Parametern Temperatur und Druck ist die Auswahl des Precursors, vor allem dessen Flüchtigkeit sowie die thermische Stabilität unter den gewählten Prozessbedingungen, entscheidend für die Durchführung.
Heterogenisierung homogener Katalysatoren
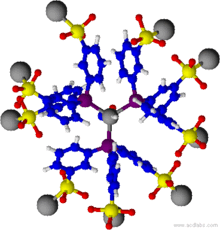 Wasserlöslichler Rhodium-Triphenylphosphantrisulfonat-Komplex (TPPTS)
Wasserlöslichler Rhodium-Triphenylphosphantrisulfonat-Komplex (TPPTS)Durch die Heterogenisierung homogener Katalysatoren wird versucht, die Vorteile der homogenen Katalyse, wie hohe Selektivität und vollständige Verfügbarkeit der katalytisch aktiven Spezies mit den Vorteilen der heterogenen Katalyse, wie der leichten Abtrennbarkeit von Katalysator und Reaktanden zu vereinigen.
Die Heterogenisierung homogener Übergansgmetallkomplexe erfolgt meist durch die Fixierung der löslichen Komplexe an einen festen Träger. Die Fixierung kann dabei durch Modifikation der Komplexliganden kovalent erfolgen, aber auch eine ionische oder adsorptive Fixierung ist möglich. Bei porösen Festkörpern ist auch die physikalische Einlagerung in die Porenstruktur des Festkörpers möglich.
Die bislang bekannten Beispiele zeigen allerdings, dass eine zu starke Fixierung zum Nachlassen der katalytischen Aktivität führt, während eine zu schwache Fixierung zum „Ausbluten“ oder „leaching“ des Komplex aus dem Festkörper führt.
Eine weitere Methode ist die Heterogenisierung in flüssig-flüssig-Systemen, wobei der Metallkomplexkatalysator durch Ligandenmodifikation in der Regel wasserlöslich wird und sich so einfach von der entstehenden organischen Phase trennen lässt. Ein bekanntes Beispiel ist die Hydroformylierung nach dem Ruhrchemie/Rhône-Poulenc-Verfahren, wobei Rhodium mit Triphenylphosphansulfonat komplexiert wird, welches durch die Ligandensubstitution mit Sulfonatgruppen hydrophile Eigenschaften besitzt. Die Reaktion findet in der wässrigen Phase statt. Die organische Produktphase wird dabei mittels Phasenabscheidung abgetrennt, die wässrige Katalysatorphase wird wieder dem Reaktor zugeführt.
Katalysatordeaktivierung und Regeneration
Die Mechanismen der Katalysatordeaktivierung sind vielfältig. Grob kann die Deaktivierung eingeteilt werden in mechanische, etwa durch Abrieb oder Zerfall, thermische wie zum Beispiel durch Sinterung, physikalische wie die Verkokung oder die physikalische Blockade aktiver Zentren sowie die chemische Deaktivierung durch Bildung inaktiver Metallkomponenten wie beispielsweise Sulfide.
In der heterogenen Katalyse sind zum Beispiel bei Raffinerieprozessen die Verkokung, die Sinterung der aktiven Oberfläche oder der Zerfall des Katalysators durch mechanischen Abrieb, zum Beispiel bei Fluid-Bed-Verfahren, bekannt. Durch Alterungsprozesse kann die katalytisch aktive Oberfläche verkleinert werden oder Poren können verstopfen, zum Beispiel in Zeolithen. Irreversibel ist oft die Deaktivierung durch Phasenumwandlungen. Diese wird beispielsweise bei Zink-/Aluminiumoxid-Katalysatoren für die Methanolsynthese beobachtet, die zu hohen Temperaturen ausgesetzt wurden. Durch Bildung einer Spinell-Phase wird der Katalysator deaktiviert und kann nicht regeneriert werden.
Zu den Regenerationsverfahren zählt beispielsweise das Abbrennen von Koks von Kontakten, die in Crack-Prozessen oder dem katalytisches Reforming eingesetzt werden oder die Oxichlorierung zur Wiederherstellung acider Zentren. Ist der Katalysator soweit deaktiviert, dass eine Regeneration nicht mehr sinnvoll ist, wird der Katalysator aus dem Prozess ausgeschleust. Bei Edelmetallkatalysatoren werden die Träger gegebenenfalls geschmolzen und das Edelmetall durch Verhüttungs- und elektrochemische Prozesse zurückgewonnen.
Analytische Verfahren zur Charakterisierung heterogener Katalysatoren
Heterogene Katalysatoren können mit den herkömmlichen Methoden zur Analyse von Festkörpern analysiert werden. Mit Methoden wie zum Beispiel der Atomabsorptionsspektroskopie oder der Röntgenfluoreszenzanalyse kann die chemische Zusammensetzung ermittelt werden. Daneben wurden spezielle Methoden zur Katalysatorcharakterisierung entwickelt, wie zum Beispiel die selektive Adsorption von Wasserstoff oder Kohlenstoffmonoxid an Metallzentren zur Bestimmung der Dispersion von Metallen. Neben der chemischen Zusammensetzung sind die Art der im Kontakt vorhandenen Phasen, die Porenstruktur, die innere Oberfläche, die Charakterisierung saurer Zentren und die Oberflächenbeschaffenheit von Interesse.
Oberflächensensitive Methoden
 Messsystem für die Röntgen-Photoelektronenspektroskopie (XPS) mit Halbkugelanalysator, Röntgenröhren und diversen Präparationsmethoden
Messsystem für die Röntgen-Photoelektronenspektroskopie (XPS) mit Halbkugelanalysator, Röntgenröhren und diversen PräparationsmethodenUm Strukturen und Vorgänge an Oberflächen zu untersuchen werden spezielle Methoden der Oberflächenanalytik verwendet. Diese detektieren nur Prozesse und Strukturen, die sich von denen des Festkörpers unterscheiden. Dazu werden die Wechselwirkungen von Elektronen, Photonen, neutralen Teilchen, Ionen oder Wärme mit den Oberflächen genutzt. Für verschiedene Fragestellungen wurde eine Vielzahl von mikroskopischen, spektroskopischen, thermoanalytischen, Adsorptions- und Beugungsmethoden entwickelt.
Eine Voraussetzung für die Oberflächensensitivität einer Methode ist oft, dass das wechselwirkende oder das detektierte Teilchen oder Welle eine geringe mittlere freie Weglänge in der Materie besitzt. Deshalb ist für viele Methoden ein Ultrahochvakuum nötig. Da viele Reaktionen aber unter höherem Druck ablaufen, ist die Übertragung der mit oberflächensensitiven Methoden gewonnen Erkenntnisse in die industrielle Praxis oft schwierig.
Bestimmung der inneren Oberfläche
Die BET-Messung (nach den Nachnamen der Entwickler des BET-Modells, Stephen Brunauer, Paul Hugh Emmett und Edward Teller) ist ein Analyseverfahren zur Bestimmung der inneren Oberfläche von porösen Festkörpern mittels Gasadsorption. Durch Ermittlung einer Adsorptions-Desorptions-Isotherme, in der Regel von Stickstoff, in bestimmten Druckbereichen ist die gemessene Menge an adsorbiertem beziehungsweise freiwerdendem Stickstoff proportional zur inneren Oberfläche. Die BET-Oberfläche wird in m2·g−1 angegeben.
Bestimmung saurer Zentren
Eine Methode zur Bestimmung der Acidität heterogener Katalysatoren ist die temperaturprogrammierte Desorption von Ammoniak. Dabei können anhand der Desorptionstemperatur Lewis- und Brønsted-Zentren unterschieden werden.
Zum Nachweis wird die Katalysatorprobe zunächst mit Ammoniak gesättigt und danach zum Beispiel im Inertgasstrom temperaturprogrammiert aufgeheizt und das desorbierte Ammoniak quantitativ erfasst. Physisorbiertes Ammoniak ohne Anlagerung an acide Zentren desorbiert bei Temperaturen zwischen 150 und 200 °C. Ammoniak, welches im Bereich zwischen 200 und 400 °C desorbiert, wird Lewis-sauren Zentren zugeschrieben. Bei höheren Temperaturen von 400 bis 600 °C desorbierendes Ammoniak stammt aus der Zersetzung vom Ammoniumionen, das sich durch Reaktion von Ammoniak mit Brønsted-Zentren gebildet hat.
Bestimmung der Metalldispersion
Die Aktivität von Trägerkatalysatoren wird durch die Dispersion des Metalls auf dem Trägermaterial bestimmt, da nur die Oberflächenatome des Metalls an der chemischen Reaktion teilnehmen können. Ein oft angewandtes Verfahren zur Messung der Dispersion der Metalle ist die Chemisorption von Wasserstoff oder von Kohlenstoffmonoxid. Dabei wird angenommen, dass das stöchiometrische Verhältnis Metall – Wasserstoff 1:1 beträgt, der Wasserstoff also dissoziativ chemisorbiert wird. Sollte der Wasserstoff im Metall löslich sein wie zum Beispiel bei Palladium-Katalysatoren, kann das Ergebnis der Wasserstoffchemisorptionsmessung fehlerbehaftet sein.
Die Bestimmung der Menge des chemisorbierten Wasserstoffs kann gravimetrisch oder volumetrisch erfolgen. Alternativ kann der chemisorbierte Wasserstoff mit Sauerstoff titriert werden und das entstandene Wasser detektiert werden.
Bestimmung der Porengrößenverteilung
Nach IUPAC werden drei Größenbereiche bei Poren unterschieden:
- Mikroporen mit einem Durchmesser von < 2 nm
- Mesoporen mit einem Durchmesser von 2 nm bis 50 nm
- Makroporen mit einem Durchmesser von > 50 nm[17]
Die Porengrößenverteilung eines heterogenen Katalysators kann mittels Quecksilberporosimetrie bestimmt werden. Die Eigenschaft von Quecksilber, sich wie eine nichtbenetzende Flüssigkeit zu verhalten ist die Grundlage dieser Methode. Dazu wird Quecksilber bei Drücken von bis zu 4.000 bar in Poren unterschiedlicher Größe gedrückt. Dabei werden anfangs die großen und bei höheren Drücken die kleineren Poren erfasst. Über die Abhängigkeit der Quecksilbermenge in Abhängigkeit vom aufgewendeten Druck können Aussagen über die Beschaffenheit, Form, Verteilung und Größe der Poren gemacht werden. Mittels Quecksilberporosimetrie können relativ große Porenbereichverteilungen ermittelt werden.
Bestimmung der Partikelgröße
Zur Bestimmung der Partikelgröße von auf Trägern dispergierten Metallpartikel können sowohl direkt abbildende Methoden wie die Transmissionselektronenmikroskopie als auch Streumethoden eingesetzt werden. Direkt abbildende Methoden erfordern in der Regel einen größeren Probenpräparations- sowie Auswertungsaufwand, um eine statistisch gesicherte Aussage über die Partikelgröße zu machen.
Zur Bestimmung der Partikelgröße von auf Trägermaterialien dispergierten Metallen im Bereich von circa 1 bis 100 nm kann die Röntgenkleinwinkelstreuung (engl.: Small Angle X-Ray Scattering, SAXS) eingesetzt werden. Dazu wird die Katalysatorprobe mit monoenergetischer Röntgenstrahlung bestrahlt und die Intensität der gestreuten Röntgenstrahlung bei kleinen Streuwinkeln ermittelt.
Kinetik der heterogenen Katalyse
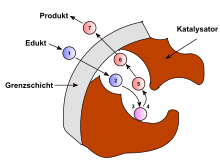 Die sieben Schritte der heterogenen Katalyse an porösen Katalysatoren
Die sieben Schritte der heterogenen Katalyse an porösen KatalysatorenDie heterogene Katalyse in einem porösem Katalysatorkorn kann in sieben Teilschritte unterteilt werden, von denen jeder Schritt geschwindigkeitsbestimmend sein kann. Der erste Schritt ist die Diffusion der Edukte zur Oberfläche des Kontakts durch die stationäre Grenzschicht. Die Dicke der Grenzschicht verändert sich mit der Strömungsgeschwindigkeit. Der zweite Schritt ist die Diffusion der Edukte in den Poren des Kontakts zum katalytisch aktiven Zentrum. Im dritten Schritt erfolgt die Adsorption der Edukte am aktiven Zentrum. Danach folgt als vierter Schritt die Reaktion der Edukte an der Oberfläche.
Danach erfolgt der Abtransport der Produkte in umgekehrter Reihenfolge. Im fünften Schritt desorbieren die Produkte vom aktiven Zentrum. Danach erfolgt die Diffusion der Produkte durch das Porensystem des Kontakts als sechster Schritt. Im siebten Schritt diffundieren die Produkte durch die Grenzschicht in den Gasstrom in den Hauptgasstrom und werden abtransportiert. Wie bei allen konsekutiven Reaktionen ist in der Regel nur der langsamste Elementarschritt geschwindigkeitsbestimmend. Hougen und Watson entwickelten einen allgemeinen Reaktionsgeschwindigkeitsansatz für heterogen katalysierte Reaktionen:
Der kinetische Term umfasst die Geschwindigkeitskonstanten des geschwindigkeitsbestimmenden Elementarschritts, der Potentialterm umfasst die Konzentrationsterme und die Reaktionsordnung und der Adsorptions- oder Hemmterm umfasst die Belegung der katalytisch aktiven Zentren. Der Exponent n steht für die an der an Elementareaktion beteiligten Zentren.
Langmuir-Hinshelwood-Mechanismus
 Irving Langmuir
Irving LangmuirDer Langmuir-Hinshelwood-Mechanismus' beschreibt eine Methode für die Darstellung der Reaktionsgeschwindigkeit einer zwei- oder mehrkomponentigen heterogenen katalysierten Reaktion als Funktion von Partialdruck oder Konzentration unter Adsorption aller Reaktanden.
Irving Langmuir untersuchte 1915 die Abnutzung der Wolfram-Glühfäden durch Sauerstoff, die zur Entwicklung einer Theorie über die Adsorption von Gasen als Funktion des Partialdrucks führte. In Zusammenhang mit der Theorie der aktiven Zentren von Hugh Stott Taylor gelang Langmuir 1927 die Formulierung der Langmuir-Hinshelwood Kinetik.[18] Bis zur Entwicklung von oberflächensensitiven Analysenmethoden in der zweiten Hälfte des zwanzigsten Jahrhunderts waren kinetische Analysen ein wichtiges Instrument zur Untersuchung heterogen-katalysierter Prozesse.
Für eine unimolekulare, heterogen-katalysierte Reaktion wird angenommen, dass die Reaktionsrate proportional dem Bedeckungsgrad Θ ist.
 sowie
sowie
Für sehr kleine Werte von bAPA << 1 ergibt sich
das heißt, die Reaktion ist erster Ordnung im Bezug auf PA.
Ist bAPA >> 1 sehr groß, ergibt sich, dass
das heißt, die Ordnung der Reaktion ist nullter Ordnung im Bezug auf PA.
Für Reaktionen zwischen zwei Molekülen basiert der Mechanismus auf der Annahme, dass zwei Reaktanden A und B auf der Oberfläche S eines heterogenen Katalysators adsorbiert werden und im adsorbierten Zustand eine bimolekulare Reaktion abläuft.[19]
Die Geschwindigkeitskonstanten für die Adsorption und Desorption von A und B und die Reaktion sind
 ,
,  ,
,  ,
,  und
und 
Mit der Konzentration der durch A und B besetzten Oberflächenplätze,
 und
und  ,
,
dem Bedeckungsgrad
 und der Anzahl der aktiven Zentren
und der Anzahl der aktiven Zentren
Mit der Definition des Bedeckungsgrades ΘA und ΘB sowie den Adsorptionskoeffizienten bA und bB
 sowie
sowie
ist das Geschwindigkeitsgesetz dann
Wenn zwei Moleküle um denselben Adsorptionsplatz konkurrieren, dann ist die Reaktionsgeschwindigkeit am größten, wenn
 .
.
Für PA = n PB ergibt sich, dass die Reaktionsgeschwindigkeit dann am größten ist, wenn PA/n = PB. Bei konstantem Partialdruck von B durchläuft die Reaktionsgeschwindigkeit als Funktion des Partialdrucks von A ein Maximum.
Eley-Rideal-Mechanismus
Beim Eley-Rideal-Mechanismus, der 1938 von D. D. Eley und E. K. Rideal vorgeschlagen wurde, adsorbiert zunächst Edukt A auf der Katalysatoroberfläche: [20]

Anschließend reagiert das adsorbierte Edukt mit einem weiteren Edukt B aus der Gasphase zum Produkt C:

Im letzten Schritt desorbiert Produkt C:

Der Eley-Rideal-Mechanismus ergibt sich aus dem Langmuir-Hinshelwood-Mechanismus als Grenzfall, wenn der Adsorptionskoeffizient von B gegen Null geht. Dann ist die Reaktionsgeschwindigkeit proportional dem Bedeckungsgrad von A und dem Partialdruck von B:
Reaktionen, die nach einem reinen Eley-Rideal-Mechanismus ablaufen, sind relativ selten und wurden bislang nur für einige Reaktionen nachgewiesen, etwa der Wasserstoff-Deuterium-Austausch. Oft sind schwach gebundene Oberflächenspezies in den Mechanismus eingebunden. Der Grund dafür sind die Reaktionszeiten. Eine Gas-Oberflächen-Kollision dauert nur einige Picosekunden, während oberflächengebundene Spezies eine Lebenszeit von einigen Microsekunden haben können.
Reaktionskinetische Untersuchungsmethoden
 Regimes der heterogenen Katalyse
Regimes der heterogenen KatalyseKinetische Untersuchungen von Gas-Feststoffreaktionen werden experimentell in Apparaten wie dem integralen Zapfstellenreaktor oder dem differenziellen Kreisgasreaktor durchgeführt. Beide Apparatetypen weisen verschiedene methodische sowie apparative Vor- und Nachteile auf und müssen für die jeweilige Untersuchung ausgelegt werden, wobei die einfachste Form der Festbettreaktor mit verschiedenen Zapfstellen darstellt.
Ziel der kinetischen Untersuchungen ist es unter anderem, den geschwindigkeitsbestimmenden Schritt der heterogen katalysierten Reaktion zu bestimmen. Dabei werden drei verschiedene Regimes unterschieden. Neben der chemischen Reaktion treten auch diffusionskontrollierte Schritte wie die Diffusion in der Grenzschicht oder die Porendiffusion als geschwindigkeitsbestimmende Schritte auf.
Integralreaktor
Der Intergralreaktor wird oft als einfach zu bauender Festbett-Rohrreaktor mit Zapfstellen ausgeführt. Entlang des Reaktors werden dazu in regelmäßigen Abständen Zapfstellen eingebaut, die eine Probennahme und damit die Bestimmung eines Konzentrationsprofils über die Reaktorlänge erlauben. Das Design entspricht oft einem industriellen Reaktor im Kleinmaßstab. Die Dimensionen sollten so gewählt werden, dass die Verwendung von industriell genutzten Katalysatoren möglich ist und damit Transportvorgänge etwa denen in der industriellen Praxis entsprechen. Ein bedeutender Nachteil ist das Auftreten von Konzentrations- und eventuell Temperaturgradienten, die eine direkte Messung der Reaktionsgeschwindigkeit verhindern. Die Aufarbeitung der Messdaten erfordert meist eine numerische Lösung von Differenzialgleichungen.
Differenzialreaktor
Als Differenzialreaktor werden Berty-Reaktoren eingesetzt. Es handelt sich dabei um einen Kreisgasreaktor, in dem eine Turbine einen Zirkulationsstrom erzeugt, wodurch die Edukte und Produkte vermischt werden. Typische Drehzahlen liegen in der Größenordnung von 4.000 bis 10.000 Umdrehungen pro Minute.
Die eingesetzte Katalysatormenge ist bei diesem Reaktor in der Regel klein. Ist der Kreislaufvolumenstroms gegenüber dem Eingangsvolumen, das so genannten Kreis- oder Rücklaufverhältnis, größer als zehn, erreicht man Bedingungen, die dem ideal durchmischten Rührkessel entsprechen. Konzentrations- beziehungsweise Temperaturgradienten treten dann nicht auf. Durch die Isothermie wird eine genaue Ermittlung der Temperaturabhängigkeit des Umsatzes möglich. Ein Nachteil sind die relativ großen Wandflächen und Toträume, die zu Blindreaktionen führen können und damit die Messergebnisse verfälschen.
Kenngrößen
Eine wichtige Kenngröße heterogenkatalytischer Prozesse ist die Raumgeschwindigkeit (englisch: Gas hourly space velocity; GHSV; bei Flüssigkeiten: liquid hourly space velocity; LHSV). Die GHSV ist der Quotient des Gasvolumenstrom und dem Katalysatorvolumen. Die GHSV wird bei gegebener notwendiger Verweilzeit der Komponenten am Katalysatorvolumen benötigt zur Berechnung der notwendigen Menge an Katalysator.
Einzelnachweise
- ↑ a b D. Steinborn: Grundlagen der metallorganischen Komplexkatalyse, 346 Seiten, Verlag Teubner B.G. GmbH (2007), ISBN 3835100882, ISBN 978-3835100886
- ↑ Döbereinersche Feuerzeuge. www.gnegel.de. Abgerufen am 5. Januar 2010.
- ↑ Catalysis: An Integrated Approach, von R. Santen, P. Van Leuwen, J. Moulijn. books.google.de. Abgerufen am 14. Dezember 2009.
- ↑ W. Ostwald, „Referat zur Arbeit F. Strohmann Über den Wärmegehalt der Bestandteile der Nahrungsmittel“ Z. phys. Chem. 15 (1894), S. 705 f.
- ↑ The History of the Contact Sulfuric Acid Process
- ↑ Wilhelm Normann - Erfinder der Fetthärtung
- ↑ Werner Abelshauser: Die BASF: eine Unternehmensgeschichte. C.H.Beck, 2002, ISBN 978-3406495267 (Eingeschränkte Vorschau in der Google Buchsuche).
- ↑ CCR Platforming Process For Motor Fuel Production
- ↑ I. Chorkendorff, J. W. Niemantsverdriet: Concepts of Modern Catalysis and Kinetics, 469 Seiten, Verlag John Wiley & Sons (2003), ISBN 3527305742, ISBN 978-3527305742, als Google-Book
- ↑ Eley-Rideal and Langmuir Hinshelwood Mechanism
- ↑ Selective Catalytic Reduction for power generating equipment
- ↑ An AIChE Mini History of John Mooney
- ↑ DKRW Selects ExxonMobil’s Methanol-to-Gasoline (MTG) Technology for Coal-to-Liquids Project
- ↑ a b J. Hagen: Chemiereaktoren: Auslegung und Simulation, 397 Seiten, Verlag Wiley-VCH Verlag GmbH & Co. KGaA (2004) ISBN 352730827X, ISBN 978-3527308279
- ↑ Die Schadstoffkonvertierung
- ↑ Pulverkatalysator für die Methanolsynthese
- ↑ Iupac Definition der Porengröße
- ↑ I. Chorkendorff, J. W. Niemantsverdriet: Concepts of Modern Catalysis and Kinetics, 469 Seiten, Verlag John Wiley & Sons (2003), ISBN 3527305742, ISBN 978-3527305742, als Google-Book
- ↑ Surface Science: Foundations of Catalysis and Nanoscience, von Kurt W. Kolasinski. books.google.de. Abgerufen am 3. Dezember 2010.
- ↑ D. D. Eley and E. K. Rideal, Parahydrogen conversion on tungsten, Nature, 1940, 146, 401-2. DOI:10.1038/146401d0
Literatur
- F. Schüth: Heterogene Katalyse. Schlüsseltechnologie der chemischen Industrie. In: Chemie in unserer Zeit. 40, Nr. 2, 2006, ISSN 0009-2851, S. 92–103, doi:10.1002/ciuz.200600374.
- M. Baerns, H. Hofmann, A. Renken: Chemische Reaktionstechnik, 2. Auflage, Georg-Thieme-Verlag, Stuttgart 1987.
- G. Emig, E. Klemm: Technische Chemie: Einführung in die Chemische Reaktionstechnik. 5. Auflage, Springer Verlag, Berlin 2005.
- B. Cornils, W. A. Herrmann, R. Schlögl, C. H. Wong: Catalysis from A to Z: A Concise Encyclopedia, 863 Seiten, Verlag Wiley-VCH (2003), ISBN 3527303731, ISBN 978-3527303731
- J. M. Thomas, W. J. Thomas: Principles and practice of heterogeneous catalysis. Wiley-VCH, 1997, ISBN 9783527292394 (Eingeschränkte Vorschau in der Google Buchsuche).
Weblinks
 Commons: Heterogene Katalyse – Sammlung von Bildern, Videos und Audiodateien
Commons: Heterogene Katalyse – Sammlung von Bildern, Videos und Audiodateien Wiktionary: Katalyse – Bedeutungserklärungen, Wortherkunft, Synonyme, Übersetzungen
Wiktionary: Katalyse – Bedeutungserklärungen, Wortherkunft, Synonyme, Übersetzungen



















Wikimedia Foundation.