- Knochenmarkkrebs
-
Klassifikation nach ICD-10 C90.1 Plasmazellenleukämie C91 Lymphatische Leukämie C92 Myeloische Leukämie C93 Monozytenleukämie C94, C95 Sonstige Leukämien ICD-10 online (WHO-Version 2006)  Erstbeschreibung einer Leukämie durch Rudolf Virchow 1845.
Erstbeschreibung einer Leukämie durch Rudolf Virchow 1845.Die Leukämie oder Hyperleukozytose (griechisch λευχαιμία leuchaimia, von λευκός, leukós - weiß und αἷμα, haima - das Blut), mitunter auch als Blutkrebs oder Leukose bezeichnet, ist eine Erkrankung des blutbildenden Systems. Sie wurde 1845 erstmals von Rudolf Virchow beschrieben, der auch den Namen geprägt hat.
Leukämien zeichnen sich durch stark vermehrte Bildung von weißen Blutkörperchen (Leukozyten) und vor allem ihrer funktionsuntüchtigen Vorstufen aus. Diese Leukämiezellen breiten sich im Knochenmark aus, verdrängen dort die übliche Blutbildung und treten in der Regel auch stark vermehrt im peripheren Blut auf. Sie können Leber, Milz, Lymphknoten und weitere Organe infiltrieren und dadurch ihre Funktion beeinträchtigen. Durch die Störung der Blutbildung kommt es zur Verminderung der normalen Blutbestandteile. Es entsteht eine Anämie durch Mangel an Sauerstoff transportierenden roten Blutkörperchen (Erythrozyten), ein Mangel an blutungsstillenden Blutplättchen (Thrombozyten) und ein Mangel an funktionstüchtigen weißen Blutkörperchen (Leukozyten).
Folgen sind Symptome wie Blässe, Schwäche, Blutungsneigung mit spontanen blauen Flecken und Petechien, Anfälligkeit für Infektionen mit Fieber sowie geschwollene Lymphknoten, Milz- und Lebervergrößerung und manchmal Knochenschmerzen. In Abhängigkeit vom Verlauf unterscheidet man akute und chronische Leukämien (vgl. Krankheitsverlauf). Akute Leukämien sind lebensbedrohliche Erkrankungen, die unbehandelt in wenigen Wochen bis Monaten zum Tode führen. Chronische Leukämien verlaufen meist über mehrere Jahre und sind im Anfangsstadium häufig symptomarm.
Inhaltsverzeichnis
Klassifikation und Diagnostik
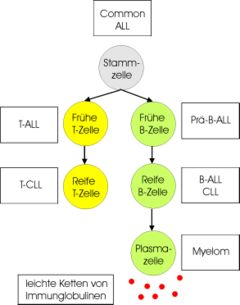 Benennung lymphatischer Leukämieerkrankungen (Rechtecke) entsprechend dem Differenzierungsstadium der entarteten Lymphozytenpopulationen (Kreise)
Benennung lymphatischer Leukämieerkrankungen (Rechtecke) entsprechend dem Differenzierungsstadium der entarteten Lymphozytenpopulationen (Kreise)Die Klassifikation der Leukämien basiert auf morphologischen und immunologischen Eigenschaften der Leukämiezellen. In den letzten Jahren gewinnen auch zunehmend zytogenetische und molekularbiologische Merkmale an Bedeutung. Je nach beteiligtem Zelltyp unterscheidet man zunächst myeloische von lymphatischen Leukämien. Myeloische Leukämien gehen von den Vorläuferzellen der Granulozyten, im weiteren Sinne auch der Erythrozyten und Thrombozyten aus, lymphatische Leukämien betreffen die Lymphozyten und ihre Vorläuferzellen.
Die wichtigsten Leukämieformen:
- akute myeloische Leukämie (AML)
- akute lymphatische Leukämie (ALL)
- chronische lymphatische Leukämie (CLL), gehört zu den niedrigmalignen Non-Hodgkin-Lymphomen
- chronische myeloische Leukämie (CML), wird zu den chronischen myeloproliferativen Erkrankungen gezählt.
Seltenere mit der CML verwandte chronische myeloproliferative Erkrankungen, die aber nicht die Kriterien einer malignen Erkrankung erfüllen, sind die
- Polycythaemia vera (PV) – hier steht die Vermehrung der Erythrozyten im Blut im Vordergrund. Es sind meist auch die anderen Zellreihen, also die Leukozyten und die Thrombozyten betroffen – und die
- essentielle Thrombozythämie (ET) – hier steht die Vermehrung der Blutplättchen und deren eventuell eingeschränkte Funktion im Vordergrund.
Die Verdachtsdiagnose ist häufig bereits aus dem Blutbild und Differentialblutbild zu stellen, die genaue Klassifikation erfordert aber meist eine Knochenmarkspunktion.
Epidemiologie und Ursachen
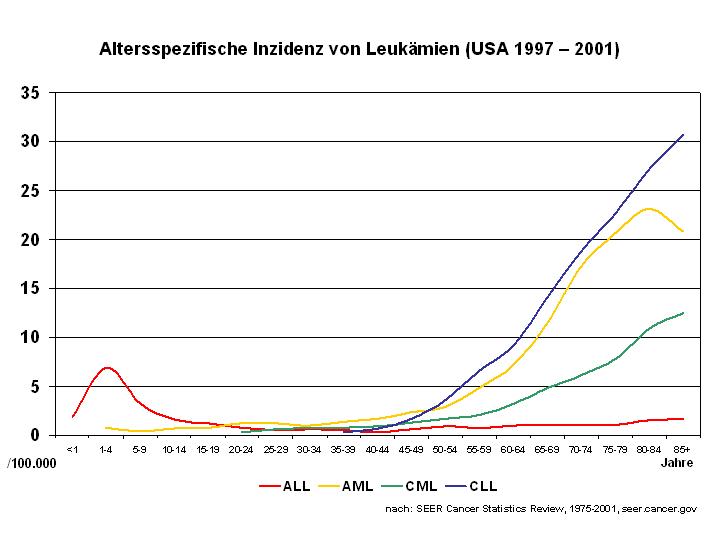
Die einzelnen Leukämietypen weisen eine typische Altersverteilung auf. Die ALL ist die häufigste Leukämie bei Kindern und kommt bei Erwachsenen seltener vor. Die AML steht bei Kindern an zweiter Stelle und ist bei Erwachsenen die häufigste akute Leukämie mit einem Altersgipfel über 60 Jahren. Die CLL tritt bei Kindern praktisch niemals auf und ist eine typische Leukämieform des älteren Menschen. Die CML ist bei Erwachsenen wesentlich häufiger als bei Kindern.
Die Ursachen von Leukämien sind noch nicht geklärt. Gerade bei akuten Formen sind die Ursachen meist unklar und können nicht in einen kausalen Zusammenhang mit pathogenen Faktoren gebracht werden. Diskutiert werden die nachfolgenden potentiell auslösenden Faktoren:
- Chemikalien, zum Beispiel Benzol,
- vorangegangene Behandlung mit Zytostatika (insbesondere Alkylanzien) aufgrund einer anderen Erkrankung (beispielsweise solide Tumore),
- ionisierende Strahlung,
- diverse Viren,
- genetische Vorbelastung
Ursachen der Leukämie bei Kindern
In Deutschland erkranken jährlich 1800 Kinder neu an Leukämie [UICC]. Auch hier sind die Ursachen weitgehend unbekannt.
Eine aktuelle Fallkontrollstudie des Kinderkrebsregisters in Mainz hat einzig eine Erkrankung mit Trisomie 21 ("Down-Syndrom") als einen starken Risikofaktor identifizieren können.[1]
Im Verdacht stehen folgende Ausgangspunkte als Risikoverursacher:
- ionisierende Strahlung
- hohes Geburtsgewicht
- Pestizidexposition
- Exposition der Mutter mit Farben, Lacken und Färbemitteln
- Sterilitätsbehandlung vor der Schwangerschaft
Die vermutete Ursache Radioaktivität für den Leukämiecluster Elbmarsch bei Hamburg sowie eine erhöhtes Risiko durch nichtionisierende Strahlung [2] ist umstritten.
Weitere Risiken lassen sich aus den Faktoren
- (junges) Alter der Mutter
- Chronische Bronchitis des Kindes
- niedriges Geburtsgewicht und
- Tonsillektomie des Kindes herleiten.
Allerdings sind die letztgenannten Zusammenhänge tendenziell durch das zu Grunde liegende Studiendesign bedingt und bedürfen einer weiteren Untersuchung. Dies gilt auch für die vermutete stark protektive Wirkung von Schutzimpfungen.
Moderat protektiv wirkt eine Atopie des Kindes/der Eltern, schwach protektiv auch das Stillen.
Die folgenden Faktoren werden im Zusammenhang mit Leukämieerkrankungen häufig diskutiert, bisher war jedoch kein Zusammenhang nachweisbar:
- Zigarettenrauchen der Eltern
- Alkoholkonsum der Mutter
- frühere Fehlgeburten
- Kaiserschnitt
- Vitamin-K-Prophylaxe
- höheres Alter der Mutter (einige Studien sehen ein schwach erhöhtes Risiko)
- Geburtsreihenfolge
- Infektionskrankheiten
- Erregerkontakte beim Kind (noch nicht ausreichend untersucht), gegenwärtig populäre Hypothese zur Erklärung der Entstehung der ALL)
- berufliche Lösungsmittelexposition der Mutter
- Röntgenuntersuchungen des Kindes
- Röntgenuntersuchungen der Mutter
- Strahlenexposition der Eltern
- Radongas in der Wohnung (für Konzentrationen kleiner 200 Bq/m³ Raumluft kein Zusammenhang, höhere Konzentrationen nicht untersucht, treten aber eher selten auf.)
- Verkehrsdichte
- Holzschutzmittel
Es ist also gegenwärtig wesentlich einfacher, Faktoren auszuschließen, die nicht für Leukämie verantwortlich sind.
Therapie
Grundlage der Behandlung von Leukämien ist die Therapie mit Zytostatika. Weitere Behandlungsprinzipien sind die Hochdosistherapie mit autologer Stammzellinfusion und die allogene Knochenmark- bzw. Stammzelltransplantation. Dazu wird ähnlich wie bei einer Bluttransfusion ein passender Knochenmarkspender benötigt. Untergeordnete Bedeutung hat die prophylaktische oder therapeutische Strahlentherapie. In den letzten Jahren haben sich neue Therapiemöglichkeiten durch die Anwendung von monoklonalen Antikörpern und neue spezifisch in die Krankheitsprozesse eingreifende Medikamente wie Imatinib und Dasatinib (zwei Tyrosinkinase-Inhibitoren) bei der CML und der Philadelphia-Chromosom-positiven ALL oder ATRA bei der Promyelozyten-Leukämie eröffnet. In der Therapie der Leukämien bestehen zwischen den einzelnen Formen erhebliche Unterschiede, die Einzelheiten der Therapie sind in den entsprechenden Artikeln dargestellt.
Leukämie bei Menschen mit Down-Syndrom (Trisomie 21)
Die akute Megakaryoblastenleukämie (Akute myeloische Leukämie, megakaryoblastischer Subtyp / AMkL) ist die Form von Leukämie, die am häufigsten bei jüngeren Kindern mit Down-Syndrom (Trisomie 21) auftritt; ihr Risiko an Leukämie zu erkranken, ist gegenüber dem Durchschnitt um das 20-fache erhöht, was eine Auftretenshäufigkeit von 1:100 bedeutet. Im Gegensatz zu nicht betroffenen Kindern sprechen Kinder mit zusätzlichem Erbmaterial des 21. Chromosoms jedoch meist besser auf eine Behandlung mittels Chemotherapie an, ihre Heilungs- und Überlebenschancen sind deutlich höher und Rückfälle seltener. Bei einer Studie wurde herausgefunden, dass für dieses Phänomen offenbar eine bestimmte Genmutation, die sogenannte GATA1-Mutation, verantwortlich ist, genauer gesagt das 40-kDA GATA1 Protein. Es bedingt eine verbesserte Wirksamkeit der Medikation. Jedoch liegt es wahrscheinlich ebenfalls an dieser Mutation, dass Kinder mit einer Trisomie 21 häufiger die Form von Leukämie bekommen, die durch die Mutation erfolgversprechender behandelt werden kann [3]. Abgesehen von dem erhöhten Leukämie-Risiko sind Menschen mit Down-Syndrom unterdurchschnittlich anfällig für andere Formen von Krebserkrankungen. In sechs unabhängig von einander durchgeführten Studien konnte erwiesen werden, dass z.B. Neuroblastome, Nephroblastome, Unterleibskrebs, Brustkrebs, Magenkrebs und Darmkrebs sehr selten auftreten: „Verglichen nach Alter und Geschlecht ist die Wahrscheinlichkeit für eine Person mit Down-Syndrom, an irgendeiner Form von Gewebekrebs zu sterben, um 50 bis 100 Mal niedriger“ als üblich [4]. Zurückgeführt werden kann dies neben dem durch das zusätzliche Erbmaterial offenbar begünstigten Schutzmechanismus des Körpers auch darauf, dass die mit der Trisomie 21 zusammenhängende Disposition für insbesondere Leukämie bekannt ist und eine Erkrankung aufgrund häufigerer Arztbesuche (z.B. wegen der Anfälligkeit für Atemwegserkrankungen) oft in sehr frühen Stadien erkannt und behandelt werden kann. Darüber hinaus leben die meisten Menschen mit Down-Syndrom deutlich gesünder, insbesondere Alkohol und Nikotin werden selten aktiv konsumiert, was das Risiko, an Krebs zu erkranken, zusätzlich senkt.
Chromosomale Translokationen bei menschlichen Leukämien
Reziproke Translokationen sind für Leukämien und Lymphome typisch, bei soliden Tumoren die Ausnahme. Generell betrachtet sind Translokationen ein Charakteristikum von ca. 3% aller Tumoren. Bei insgesamt 14000 verschiedenen karyotypischen Veränderungen bei Tumoren sind über 100 recurrente Translokationen beschrieben worden (Mitelman 91). Chromosomale Veränderungen bei hämatologischen Erkrankungen sind häufig und vielfältig. Ein tabellarischer Überblick soll zunächst einen Eindruck von der Vielfalt der Phänomene geben. Dabei werden zunächst Chromosomen-Translokationen und sodann Chromosomen-Deletionen angeführt. Der weitere Artikel gliedert sich in drei Teile. Zunächst werden die chromosomalen Veränderungen bei myeloischen Leukämien besprochen. Im darauffolgenden Abschnitt werden die lymphatischen Leukämien dargestellt und eine beispielhafte Bruchpunkt-Untersuchung vorgestellt. Zum Abschluss wird noch kurz auf das Burkitt-Lymphom eingegangen.
Oncogene bei Leukämien – Translokationen Protein-Klasse Oncogen Translokation Tumor Häufigkeit Tyrosin-Kinasen c-abl/bcr t(9;22)(q34;q11) CML 95% c-abl/bcr t(9;22)(q34;q11) ALL 10% axl t(;)(;) CML ?% TF myc/Ig-Gene t(8;14)(q24;q32) BL 100% pre-B-ALL 10% T-ALL 10% E2A/PBX t(1;19)(q23;p13) pre-B-ALL 10% E2A/HLF t(17;19)(q22;p13) pre-B-ALL 10% Tal-1/TCR t(1;14)(p32;q11) T-ALL 20% Tal-1/SIL t(1;)(p32;) T-ALL 20% Tal-2/TCR t(7;9)(q35;p13) T-ALL 10% Lyl-1/TCR t(7;19)(q35;p13) T-ALL 5% Ttg-1/TCR t(11;14)(pls;q11) T-ALL 10% Ttg-2/TCR t(11;14)(p13;q11) T-ALL 10% HD-Gene: Hox-11/TCR t(10;14)(q24;q11) T-ALL 7% HRX t(11q23) Multilinage ?% Rezeptoren: RARA/PML t(15;17)(q21;q21) PML 100% bcl-Gene: bcl-1/Ig t(11;14)(q32;q21) CentroCyt 30% CLL 3% bcl-2/Ig t(14;18)(q13;q32) Foll Diff 20% CLL 5% bcl-3/Ig t(14;19)(q32;q13) CLL Andere: DEK/CAN t(6;9)(p23;q34) AML/MDS SET/CAN t(;)(;) AML,MDS MLL t(11q23) AML,ALL TAN-1 t(7;9)(q34;q34.3) T-ALL 42% AML-1 t(8;21) AML IL-3 t(5;14)(q31;q32) pre-B-ALL Die folgende Tabelle gibt einen Überblick über die chromosomalen Deletionen bei verschiedenen menschlichen Leukämien.
Oncogene bei Leukämien – Deletionen Proteinklasse Tumor Häufigkeit ras-Gene AML 50% ALL 15% CML 5% p53 CML 20% AML 3-7% pre B-ALL 2% T-ALL 2% BL 30% CLL 15% RB-1 Ph1+-ALL 30% AML 3% AMML 25% T-ALL 20% WT-1 AML 20% Chromosomale Translokationen bei chronischer myeloischer Leukämie (CML)
Bei der chronischen myeloischen Leukämie kommt es in 95% aller bisher untersuchten Fälle durch eine chromosomale Translokation zu einer Fusion des c-abl Gens auf dem Chromosom 9q34 mit dem bcr-Gen auf dem Chromosom 22q11 mit dem Ergebnis eines alterierten Chromosoms, dem Philadelphia-Chromosom, und der Expression eines chimärischen Proteins, dem abl/bcr-Produkt, das in drei Varianten als p190, p210 und p230 vorkommt und Tyrosinkinaseaktivität aufweist. Das Fusionsprotein resultiert in einer konstitutiven Aktivierung der abl-Tyrosinkinase und stimuliert vielfältige Signalwege, z.B. p21 Ras, PI3 Kinase, Jun, myc.
Chromosomale Translokationen bei akuter myeloischer Leukämie (AML)
Bei den akuten myeloischen Leukämien findet sich eine Vielzahl unterschiedlicher Mutationen. Bei der AML finden sich in bis zu 50% der untersuchten Fälle Mutationen im N-ras Lokus, in ca 5% der untersuchten Fälle Mutationen in p53, in weniger als 3% der untersuchten Fälle Mutationen im RB-1 Gen und in ca 20% Veränderungen im WT-1 Lokus. Vereinzelt sind Fusionen von SET/CAN, DEK/CAN, MLL und AML-1 Genen beschrieben worden. Im Folgenden werden die beteiligten Oncogene näher charakterisiert.
Chromosomale Translokationen bei anderen myeloischen Leukämien
Bei der akuten myelomonocytische Leukämie (AMML) finden sich häufig Mutationen im RB-1 Lokus. Eine Besonderheit bei den AML stellt die Promyelozytenleukämie dar, bei der in 100% der untersuchten Fälle eine Translokation t(15;17) (q21;q21) beschrieben ist mit dem Ergebnis einer Fusion von PML und RARa. Das Humane Trithorax-Homolog findet sich auf dem Chromosom 11q23. Die HRX-Translokationen findet sich bei biphänotypischen Leukämien. Trithorax ist ALL-1.
Chromosomale Translokationen bei T-Zell-Leukämien
Die folgende Tabelle gibt eine Übersicht über das Vorkommen von Translokationen bei akuten T-Zell-Leukämien.
Translokationen bei T-Zelleukämien t(8;14)(q24;q11) c-myc 8q24 TCR-alpha/delta 14q11 t(7;19)(q35;p13) TCR-beta 7q35 Lyl-1 19p13 t(1;14)(p32;q11) Tal-1(Scl,Tcl-5) 1p32 TCR-alpha/delta 14q11 t(7;9)(q35;q34) TCR-beta 7q35 Tal-2 9q34 t(11;14)(pl5;q11) Rhom-1(Ttg-1) 11p15 TCR-alpha/delta 14q11 t(11;14)(p13;q11) Rhom-2(Ttg-2) 11p13 TCR-alpha/delta 14q11 t(7;11)(q35;p13) TCR-beta 7q35 Rhom-2 11p13 t(10;14)(q24;q11) Hox-11(Tcl-3) 10q24 TCR-alpha/delta 14q11 t(7;10)(q35;q24) TCR-beta 7q35 Hox-11 10q24 t(7;9)(q34;q34.3) TCR-beta 7q34 Tan-1 9q34.3 Interessanterweise sind alle betroffenen proto-oncogene Transkriptionsfaktoren: c-myc, Lyl-1, Tal-1,2 sind helix-loop-helix-Proteine; Rhom-1,2 (Ttg-1,2) sind LIM-Domaine-Proteine, Hox-11 (Tcl-3) ist ein Homeoboxgen und Tan-1 ein notch-Homolog. Involviert sind jeweils immer TCR-beta oder TCR-alpha/delta. Vergleichsweise konsistente Mutationen in T-ALLs finden sich auch bei p53 Jonveaux und im RB-Lokus Ahuja und Ginsberg allerdings ohne Translokationen in den Bereich rearrangierender Loci. Untersucht man die verschiedenen Loci, so findet man folgende Verteilung der translocierenden Regionen.
In die TCR-alpha/delta-Region = 14q11 translocieren:Chromosomale Lokalisation TCR-alpha/delta-translocierender Oncogene c-myc 8q24 Tal-1 1p32 Rhom-1 11p15 Rhom-2 11p13 Hox-11 10q24 In die TCR-beta-Region = 7q35 translocieren:
Chromosomale Lokalisation TCR-beta-translocierender Oncogene Lyl-1 19p13 Tal-2 9q34 Rhom-2 11p13 Hox-11 10q24 Tan-1 9q34.3 Die aufgelisteten Translokationen bei T-ALL haben eine Reihe von Gemeinsamkeiten. Es sind jeweils zwei typische codierende Regionen betroffen: TCR-Gene und Transkriptionsfaktoren. Stets ist das betroffene Allel des TCR als Strukturgen zerstört und das betroffene Allel des Transkriptionsfaktors als Strukturgen intakt, in seiner Regulation aber gestört. Meistens sind die betroffenen Transkriptionsfaktoren zellinienfremde Gene. Üblicherweise wird ihre Funktion im Rahmen der Zelldifferenzierung vermutet. Im Bereich von 11p13 sind die Bruchpunkte unabhängig vom translocierenden Partnerchromosom in einem kleinen Bereich geclustert. Außerdem finden die Translokationen bei unreifen Zellen statt, so dass man schlussfolgern muss, dass eine aberrante Expression von an der Zelldifferenzierung von nichtlymphatischem Gewebe beteiligten Transkriptionsfaktoren in primitivem lymphoiden Gewebe einen wesentlichen Anteil an der malignen Transformation haben kann.
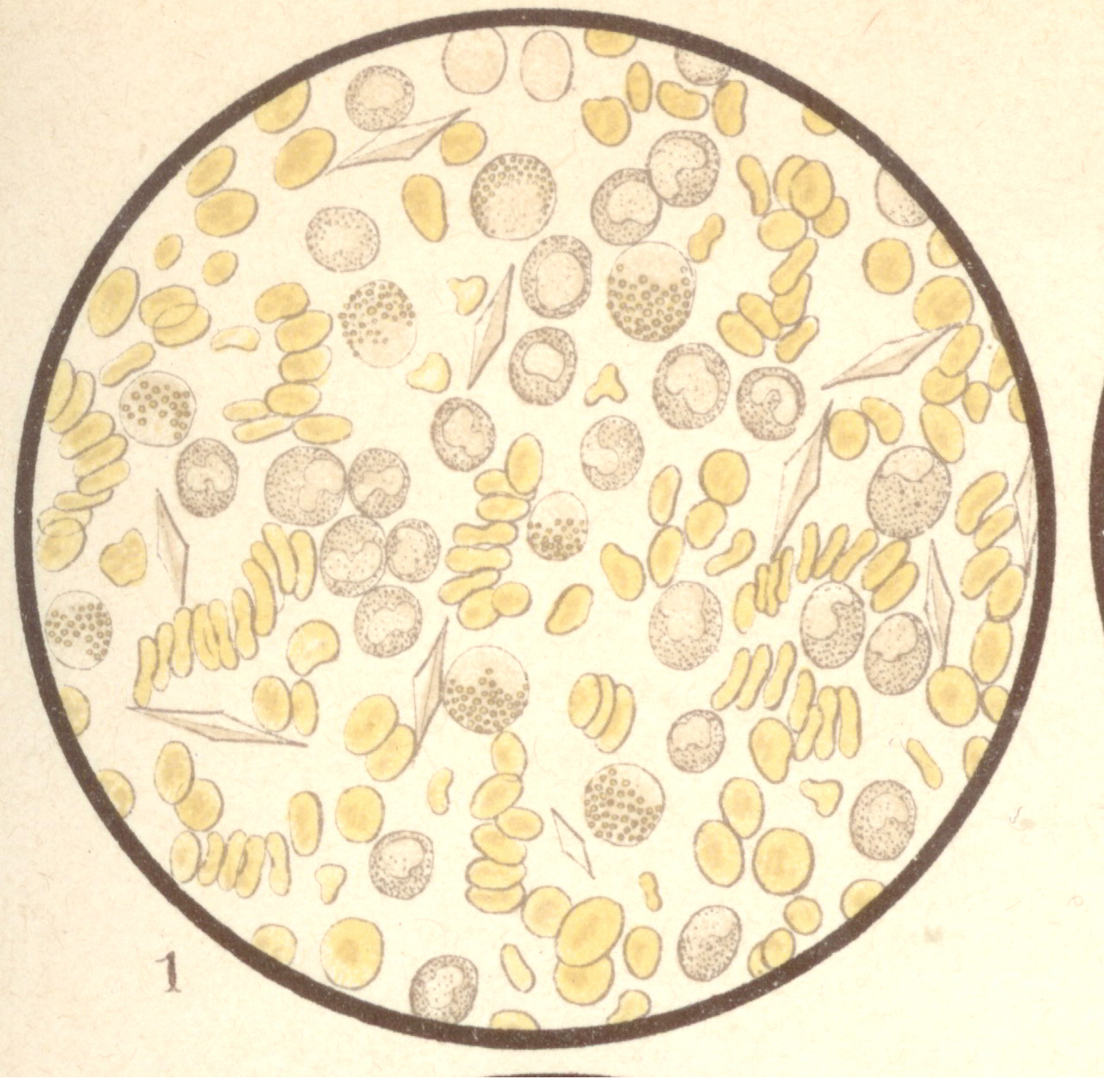
Aus Lehmann's Medicin Handatlanten Band XV. von 1897
 Bild 1: Markzellen (Leukaemia lieno-medullaris).
Bild 1: Markzellen (Leukaemia lieno-medullaris).(Lehmann's Medicin Handatlanten Band XV. - Atlas der Klinischen Untersuchungsmethoden nebst Grundriss der klinischen Diagnostik und der speziellen Pathologie und Therapie der inneren Krankheiten, von Dr. Christfried Jakob, 2. unveränderte Aufl., München 1897, Verlag von J.F.Lehmann
Bild 1: Markzellen (Leukaemia lieno-medullaris). Diese Zellen finden sich im normalen Blut gewöhnlich nicht, treten aber bei manchen Leukaemieformen sehr zahlreich auf. Sie haben stets einen großen, gelappten, meist nicht sehr intensiv sich färbenden Kern und meist neutrophile Körnelung. manche enthalten auch eosinophile Granula. Sie entstammen dem Knochenmark und zeigen entgegen den übrigen Leukocyten keine amöboide Bewegung. - aus "Atlas der klin. Untersuchungsmethoden, 1897"
 Bild 2: Markzellen in Kernteilung.
Bild 2: Markzellen in Kernteilung.Bild 2: Markzellen in Kernteilung. von einem Fall mit Leukaemie. Es fanden sich die verschiedenen Stadien der indirekten Karyokinese (Sternform, Abschnürung, Knäuelform). Vergrößerung 1000-fach. Haematoxylinfärbung. - aus "Atlas der klin. Untersuchungsmethoden, 1897"
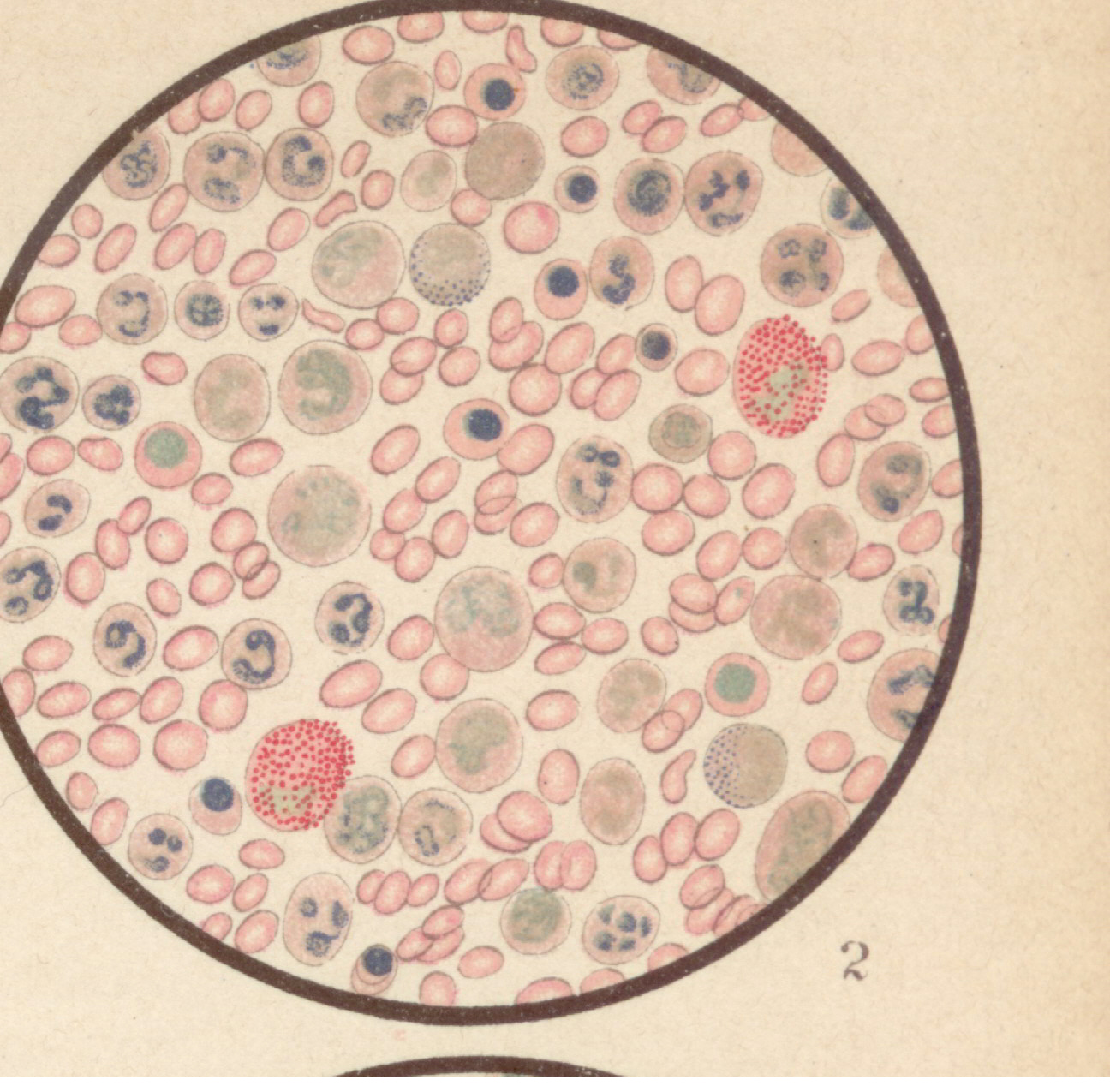
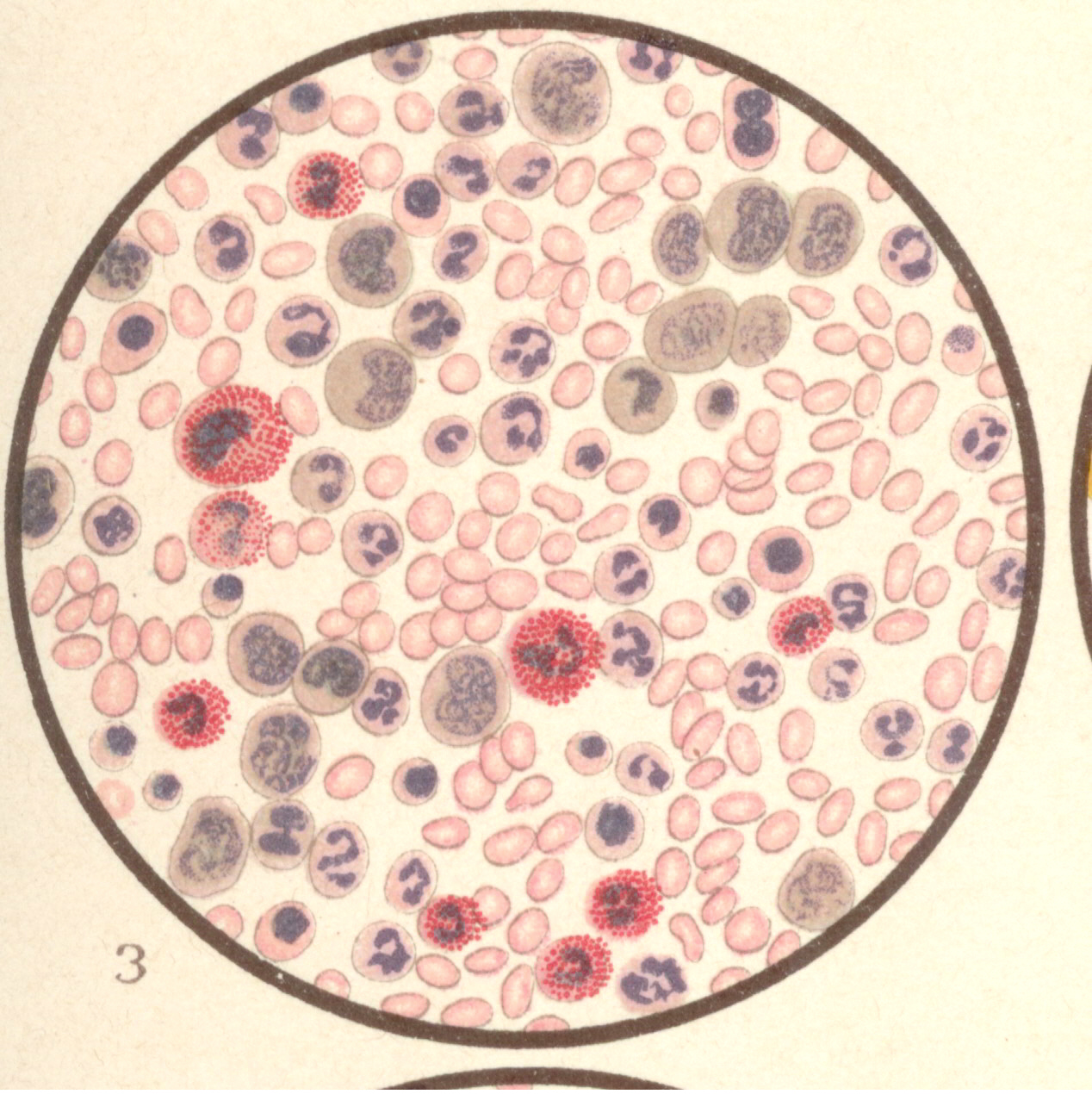
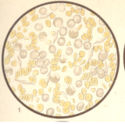 Bild 3: Frisches Blutpräparat aus einer leukämischen Leiche.
Bild 3: Frisches Blutpräparat aus einer leukämischen Leiche.Bild 3: Frisches Blutpräparat aus einer leukämischen Leiche. Die weißen Blutkörperchen sind enorm vermehrt (Zählung bei Lebzeiten ergab 360,000 weiße Blutkörperchen im Kubikmilimeter und 2,600,000 rote Blutkörperchen). Man erkennt deutlich die fein und grobkörnig (α-Granula) granulierten Leukocyten; bei Essigsäurezusatz erkennt man auch die Kerne gut; es finden sich viele mononukleäre (besonders große Formen). Außerdem haben sich im Leichenblut die "Charcot-Leydenschen Kristalle" zahlreich ausgebildet (im Leben sind sie nicht zu sehen). - aus "Atlas der klin. Untersuchungsmethoden, 1897"
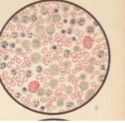 Bild 4: Gefärbtes Präparat bei Leukaemia lieno-medullaris.
Bild 4: Gefärbtes Präparat bei Leukaemia lieno-medullaris.Bild 4: Gefärbtes Präparat bei Leukaemia lieno-medullaris. Methylenblau - Eosinfärbung. Die roten Blutkörperchen sind rot, die Kerne blau, die α-Granula rot, die basophilen blau. Vergrößerung 300-fach. - aus "Atlas der klin. Untersuchungsmethoden, 1897"
 Bild 5: Gefärbtes Präparat bei Leukaemia lieno-medullaris.
Bild 5: Gefärbtes Präparat bei Leukaemia lieno-medullaris.Bild 5: Gefärbtes Präparat bei Leukaemia lieno-medullaris. Haematoxylin - Eosinfärbung. Die roten Blutkörperchen sind rot, die Kerne blau, die α-Granula rot, die basophilen blau. Vergrößerung 300-fach. - aus "Atlas der klin. Untersuchungsmethoden, 1897"
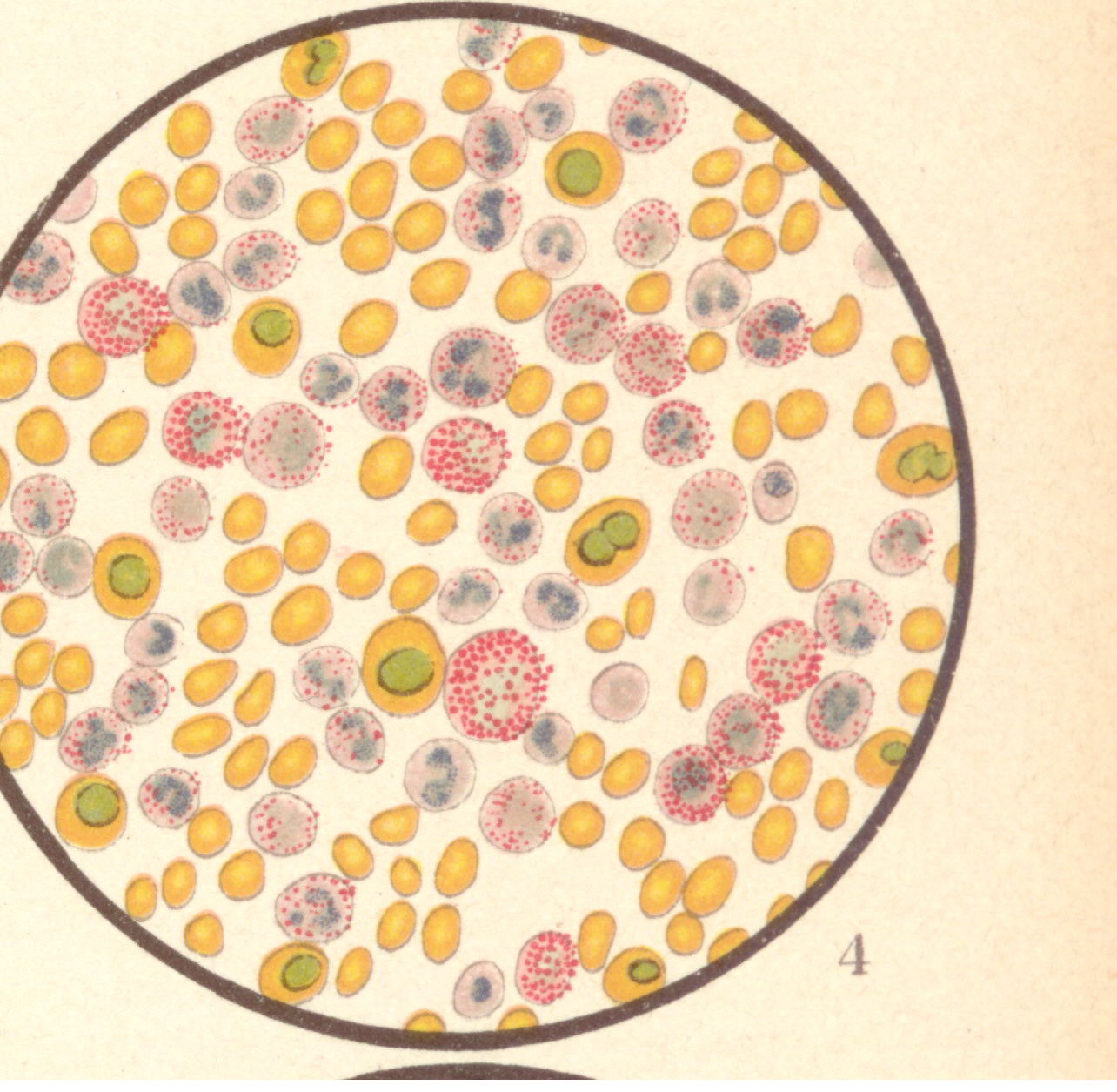
 Bild 6: Gefärbtes Präparat bei Leukaemia lieno-medullaris.
Bild 6: Gefärbtes Präparat bei Leukaemia lieno-medullaris.Bild 6: Gefärbtes Präparat bei Leukaemia lieno-medullaris. Triacidfärbung. Die roten Blutkörperchen sind gelb, ihre Kerne grün, die Leukocytenkerne bläulich, die α-Granula rot, die neutrophilen Granula violett. Es findet sich eine mäßige Poikilocytose; die weißen Blutkörperchen sind stark vermehrt. An der Vermehrung nehmen teil die polynukleären neutrophilen und besonders die eosinophilen Zellen; sodann aber finden sich (im Gegensatz zu Leukocytose) die großen mononukleären Markzellen zum Teil mit α-Körnung. Es finden sich kernhaltige rote Blutkörperchen. Vergrößerung 300-fach. - aus "Atlas der klin. Untersuchungsmethoden, 1897"
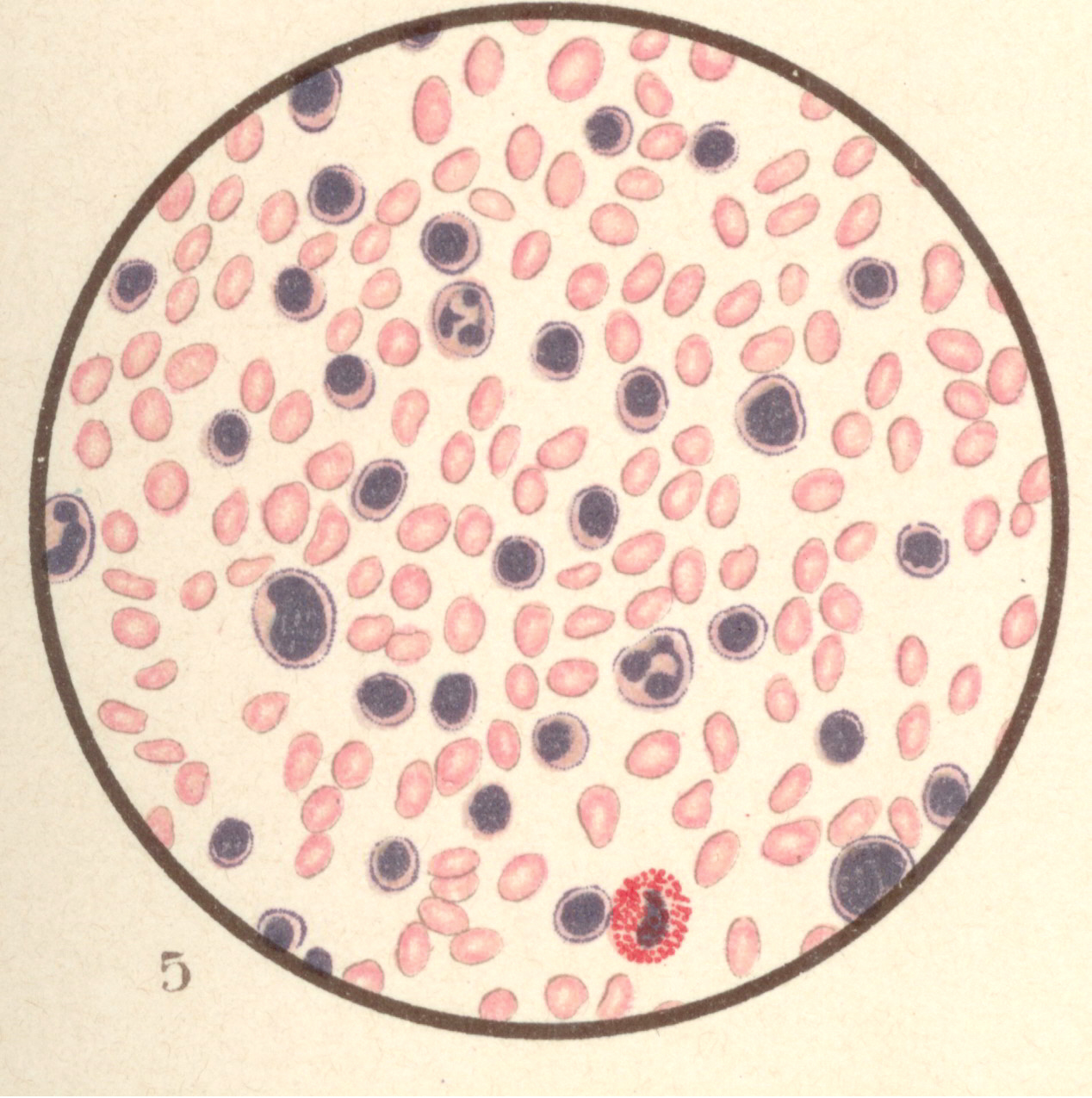
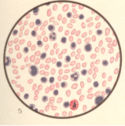 Bild 7: Leukaemia lymphatica.
Bild 7: Leukaemia lymphatica.Bild 7: Leukaemia lymphatica. (Haematoxylin - Eosinfärbung.) Hier finden sich besonders die kleinen, mononukleären Formen stark vermehrt. Eosinophile und Markzellen, sowie kernhaltige rote Blutkörperchen seltener. - aus "Atlas der klin. Untersuchungsmethoden, 1897"
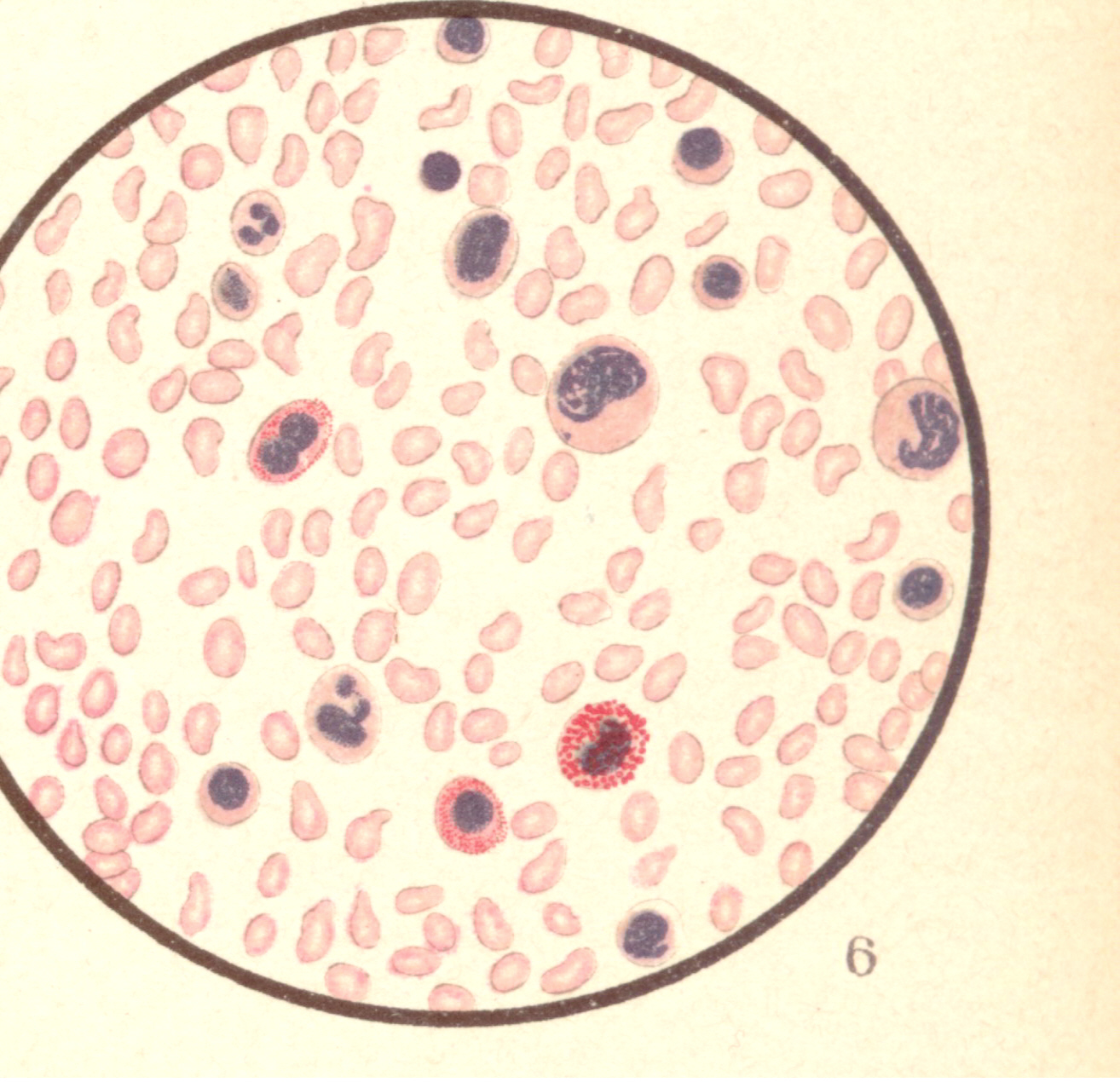
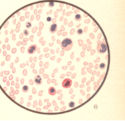 Bild 8: Akute Leukaemie bei 1/2 jährigem Kind.
Bild 8: Akute Leukaemie bei 1/2 jährigem Kind.Bild 8: Akute Leukaemie bei 1/2 jährigem Kinde. Die Zahl der weissen Blutkörperchen betrug nur 48,000 im Kubikmilimeter, trotzdem beweist der mikroskopische Befund (kernhaltige rote, etwas vermehrte mononukleäre und eosinophile Zellen ) leukämisches Blut. (Klinisch bestand großer Milztumor.) - aus "Atlas der klin. Untersuchungsmethoden, 1897"
Quellen
- ↑ Deutsches Ärzteblatt: Archiv "Ursachen von Leukämien im Kindesalter: Resümee einer Fallkontrollstudie des Deutschen Kinderkrebsregisters" (23. September 2005)
- ↑ http://www.ralf-woelfle.de/elektrosmog/allgemein/kinder.htm
- ↑ Journal of the National Cancer Institute, Februar 2005, Seite 226ff
- ↑ Zeitschrift Leben mit Down Syndrom, HG: Deutsches Down-Syndrom InfoCenter, Nr. 49, 2005, Seite 20
Siehe auch
Literatur
- Michael Begemann; Monika Begemann-Deppe: Leben mit Leukämie. Trias, Stuttgart 2000, ISBN 3-89373-568-2 (Ratgeber)
- Hermann Delbrück: Chronische Leukämien. Rat und Hilfe für Betroffene und Angehörige. 2. Auflage. Kohlhammer, Stuttgart 2004, ISBN 3-17-018369-9 (Ratgeber)
- Martin Ehrlich: Ueber Leukämie. Dissertation, Dorpat 1862 (Digitalisat als PDF)
- Nicola Gökbuget: Akute lymphatische Leukämie. UniMed-Verlag, Bremen, 1. Auflage 2007, ISBN 978-3-89599-218-6 (Fachbuch)
Weblinks
- Kompetenznetz „Akute und chronische Leukämien“ Deutsches Expertennetzwerk von Ärzten und Wissenschaftlern mit Schwerpunkt im Bereich der Leukämieforschung.
- "European Leukemia Net" Unabhängige EU-geförderte Organisation von Ärzten, Wissenschaftlern und Patienten mit Interesse an Leukämie
- Epidemiologische Studie zu Kinderkrebs (u.a. Leukämie) in der Umgebung von Kernkraftwerken - im Auftrag des Bundesamtes für Strahlenschutz 2007 - pdf 7 MB
- Audio-Beitrag zu akuten Leukämien

Bitte beachte den Hinweis zu Gesundheitsthemen!




Wikimedia Foundation.