- Parkinson-Krankheit
-
Klassifikation nach ICD-10 G20 Primäres Parkinson-Syndrom ICD-10 online (WHO-Version 2011) 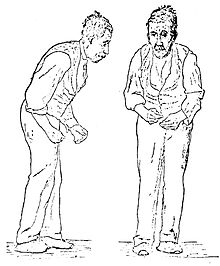 Illustration der Parkinson-Krankheit von Sir William Richard Gowers aus A Manual of Diseases of the Nervous System (Handbuch für Krankheiten des Nervensystems) von 1886
Illustration der Parkinson-Krankheit von Sir William Richard Gowers aus A Manual of Diseases of the Nervous System (Handbuch für Krankheiten des Nervensystems) von 1886
Die Parkinson-Krankheit bzw. Morbus Parkinson (weitere Synonyme: Idiopathisches Parkinson-Syndrom (IPS), Parkinsonsche Krankheit, ältere Bezeichnung: Paralysis agitans für „Schüttel-/Zitterlähmung“) ist eine langsam fortschreitende neurologische Erkrankung. Sie zählt zu den degenerativen Erkrankungen des extrapyramidal-motorischen Systems. Der Morbus Parkinson ist gekennzeichnet durch das vornehmliche Absterben von Nervenzellen in der Substantia nigra (einer Struktur im Mittelhirn) mit dem Botenstoff Dopamin. Der Mangel an Dopamin führt letztlich zu einer Verminderung der aktivierenden Wirkung der Basalganglien auf die Großhirnrinde.
Die Leitsymptome (auch Kardinal- oder Kernsymptome genannt) sind
- Rigor (Muskelstarre),
- Bradykinese (verlangsamte Bewegungen), welche bis hin zu Akinese (Bewegungslosigkeit) führen kann,
- Tremor (Muskelzittern) sowie
- posturale Instabilität (Haltungsinstabilität).
Die aktuelle Definition des Parkinson-Syndroms fordert, dass das Kardinalsymptom Brady- bzw. Akinese mit wenigstens einem der anderen Symptome (Rigor, Tremor oder posturale Instabilität) in Kombination auftritt. Daneben sind verschiedene sensible, vegetative, psychische und kognitive Störungen möglich.
Inhaltsverzeichnis
Einordnung
Der Begriff Parkinson-Syndrom ist ein Oberbegriff für Erkrankungen mit den oben genannten Leitsymptomen. Die wichtigste Erkrankung ist der hier behandelte Morbus Parkinson, eine idiopathische Erkrankung (das heißt ohne bekannte äußere oder genetische Ursache). Liegt indes eine bestimmbare äußere Ursache zugrunde, spricht man von einem sekundären oder symptomatischen Parkinson-Syndrom. Liegt ein neurodegeneratives Krankheitsbild mit anderem Schädigungsmuster mit zum Teil auch weiteren Symptomen vor, so spricht man von atypischen Parkinson-Syndromen.
Somit ergibt sich folgende Einteilung der Parkinson-Syndrome:
- das idiopathische Parkinson-Syndrom (IPS), Gegenstand dieses Artikels
- mit ca. 75 % häufigstes Parkinson-Syndrom
- das familiäre Parkinson-Syndrom
- genetisch bedingte, vererbbare Formen, selten, benannt nach jeweiligem Genort (z. B. PARK1 usw.)
- symptomatische (sekundäre) Parkinson-Syndrome
- medikamenteninduziert (z. B. bei Neuroleptika mit Dopamin-Antagonismus = Parkinsonoid), außerdem verdichten sich Hinweise, dass Amphetamingebrauch das Risiko, zu erkranken, deutlich erhöht[1]
- vaskuläres Parkinsonsyndrom, z. B. bei der zerebralen Mikroangiopathie (Morbus Binswanger)
- posttraumatisch (z. B. Boxer-Enzephalopathie)
- toxininduziert (z. B. durch Kohlenmonoxid, Mangan, MPTP)
- entzündlich (z. B. nach Enzephalitis Economo, auch bei diffusen erregerbedingten Gehirnerkrankungen wie der fortgeschrittenen HIV-Enzephalopathie)
- metabolisch (z. B. beim Morbus Wilson)
- Parkinson-Syndrome im Rahmen anderer neurodegenerativer Erkrankungen (atypische Parkinson-Syndrome)
Geschichte
Die Erkrankung wurde erstmals vom englischen Arzt James Parkinson im Jahre 1817 in der Monographie An Essay on the Shaking Palsy (Eine Abhandlung über die Schüttellähmung) beschrieben.[2] Bereits Parkinson wies auf das langsame Fortschreiten der Erkrankung hin.
Erkrankungsalter und Häufigkeit
Die Erkrankung beginnt meist zwischen dem 50. und 60. Lebensjahr (Gipfel 58. bis 62. Lebensjahr). Ein Parkinson-Syndrom kann selten bereits vor dem 40. Lebensjahr auftreten. In der Altersgruppe 40 bis 44 Jahre ist etwa einer von 10.000 Menschen betroffen. Die Manifestationsrate der Erkrankung steigt mit zunehmendem Alter bis etwa zum 75. Lebensjahr an, dann nimmt sie wieder ab. Von den über 80-Jährigen erkranken etwa 1,5–2,0 Prozent an einem Parkinson-Syndrom. In Deutschland wird derzeit von 300.000–400.000 erkrankten Menschen ausgegangen.
Ursachen
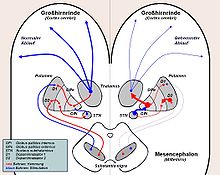 Dopaminerge Projektionen beim gesunden Menschen (links) und beim Morbus-Parkinson-Patienten (rechts); rote Pfeile stehen für Hemmung und blaue für Stimulation der Zielstruktur
Dopaminerge Projektionen beim gesunden Menschen (links) und beim Morbus-Parkinson-Patienten (rechts); rote Pfeile stehen für Hemmung und blaue für Stimulation der ZielstrukturFunktionelle Ebene: Dopaminmangel
Der Morbus Parkinson ist eine degenerative Erkrankung des Extrapyramidalmotorischen Systems (EPS) bzw. der Basalganglien. Dabei kommt es zu einem Absterben von Nervenzellen in der pars compacta der Substantia nigra (auch Nucleus niger, „schwarzer Kern“), die Dopamin herstellen und durch ihre Axone in das Putamen transportieren. Erste Krankheitszeichen fallen erst auf, wenn ca. 70 % dieser dopaminergen Zellen abgestorben sind.
Der Dopaminmangel führt über zwei Wege (siehe Bild) letztlich zu einem Ungleichgewicht in der Funktion der Basalganglien. Der Botenstoff Glutamat liegt dabei relativ im Überschuss vor. Dabei hemmt letztlich der Globus pallidus internus die motorische Aktivierung der Hirnrinde durch den Thalamus. Dies führt zu den Hauptsymptomen Rigor, Tremor und Hypokinese (siehe unten), aber auch zur Verlangsamung der geistigen Prozesse (Bradyphrenie).
Neben dem Dopaminmangel wurden auch Veränderungen anderer Neurotransmitter festgestellt. So zeigte sich in einigen Regionen des Hirnstammes ein Serotonin-, Acetylcholin- und Noradrenalin-Mangel.
Zelluläre Ebene
Die Forschung in den vergangenen zehn Jahren und die Identifizierung der vererbten Formen haben gezeigt, dass es sich beim Morbus Parkinson (MP) nicht um eine einheitliche Erkrankung handelt, sondern um eine heterogene Gruppe von Erkrankungen mit einem Spektrum von klinischen und pathologischen Ausprägungen (PARK1 bis PARK13). Monogene Formen des MP sind für etwa fünf bis zehn Prozent aller Patienten mit MP verantwortlich. Unter diesen sind Punktmutationen des alpha-Synuclein Gens (SNCA-Gen, PARK1) von besonderem Interesse, da alpha-Synuclein (SNCA) die Hauptkomponente der Lewy-Körper bei familiärem und sporadischem MP darstellt [Spillantini, 1997]. Der PARK1 Lokus wurde in einer großen Familie mit dominant vererbten MP- und Lewy-Körperchen Pathologie kartiert; zwei weitere Punktmutationen mit hoher Penetranz wurden in großen Familien identifiziert, aber nicht bei Patienten mit sporadischem MP [Polymeropoulos, 1997; Kruger, 1998; Zarranz, 2004; Berg, 2005]. Wie der Name impliziert, wurde SNCA initial als synaptisches und nukleäres Protein identifiziert [Maroteaux, 1988]. Trotz intensiver Studien ist die genaue Rolle von SNCA noch nicht klar definiert: es gibt Anzeichen dafür, dass SNCA bei der Aufrechterhaltung des synaptischen Vesikel-Pools eine Rolle spielt und Dopaminfreisetzung moduliert, aber SNCA knockout-Mäuse haben keinen offensichtlichen Phänotyp [Abeliovich, 2000; Murphy, 2000]. Interessanterweise wurden kürzlich, möglicherweise als Ausdruck einer frühen synaptischen Störung, SNCA-Aggregate in der präsynaptischen Fraktion aus menschlichem Hirngewebe identifiziert, wobei die genaue Beziehung zwischen Aggregation, zellulärer Dysfunktion und Zelltod bislang nicht bekannt ist [Kramer, 2007]. Neben Veränderungen der Aminosäuresequenz führen aber auch Duplikationen und Triplikationen zu einer Zunahme der Tendenz des Proteins Oligomere und fibrilläre Aggregate zu bilden, sodass der Regulation der SNCA-Expression und -Translation eine wichtige, zumindest modulierende Bedeutung zukommt [Singleton, 2003].
Veränderungen in regulatorischen Regionen des SNCA-Gens könnten auch mit einem höheren Risiko für die Entwicklung des MP einhergehen. Mehrere Studien haben Assoziationen von verschiedenen genetischen Varianten (single nucleotide polymorphisms; SNP) in der Promoterregion und anderen Sequenzabschnitten des SNCA-Gens mit sporadischem MP gefunden (PD Gene Database). Jüngste Untersuchungen legen eine mögliche Assoziation einzelner SNP auch in der nicht kodierenden Sequenz mit dem SNCA Expressionsniveau nahe [Fuchs, 2008].
Symptome
Die Erkrankung beginnt schleichend und schreitet danach zeitlebens fort, die Symptome werden im Verlauf stärker und daher auch besser erkennbar. Das IPS beginnt typischerweise einseitig (und bleibt im Verlauf einseitig stärker); als Frühzeichen gilt z. B. das reduzierte und später fehlende Mitschwingen eines Armes beim Laufen. Nicht selten treten Schulterschmerzen und einseitige Muskelverspannungen auf, die den Patienten zuerst zum Orthopäden führen.
Kardinalsymptome
Das Parkinson-Syndrom ist definiert durch das Vorliegen von Brady- bzw. Akinese und eines der drei anderen Leitsymptome (Rigor, Tremor, posturale Instabilität).[3]
Akinese (auch Bradykinese oder Hypokinese)
Diese allgemeine Bewegungsarmut ist Voraussetzung für die Diagnose eines Parkinson-Syndroms. Sie macht sich bei allen Bewegungen bemerkbar. So vermindert sich das Muskelspiel, was den Gesichtsausdruck bestimmt (Maskengesicht, Hypomimie), das Sprechen wird leise und undeutlich (Mikrophonie), das Schlucken verzögert sich (scheinbar vermehrter Speichelfluss – Pseudohypersalivation), die Geschicklichkeit der Hände lässt besonders bei schnellen Bewegungen nach (Schriftbild wird kleiner – Mikrographie), die Rumpfbewegungen sind erschwert (vermindertes Umlagern im Schlaf), das Gangbild wird kleinschrittig und schlurfend.
Zu diesem obligaten Krankheitszeichen muss mindestens eines der folgenden drei Symptome kommen:
Rigor (auch Rigidität)
Damit wird eine Muskelsteifheit aufgrund einer Steigerung des Muskeltonus bezeichnet. Sie wird durch eine unwillkürliche Anspannung der gesamten quergestreiften Muskulatur hervorgerufen und führt oft auch zu Muskelschmerzen. Nach außen sichtbar sind eine leichte Beugung von Ellenbogengelenk, Rumpf und Nacken sowie später der Kniegelenke. Bei passiver Bewegung der Gelenke von oberer und unterer Extremität tritt das so genannte Zahnradphänomen auf. Körpernahe Muskelgruppen sind oft stärker betroffen (axialer Rigor). Eine gekrümmte Fehlhaltung des Körperstammes durch die Tonuserhöhung wird als Kamptokormie bezeichnet.
Ruhetremor
Durch wechselseitige Anspannung gegenwirkender Muskeln entsteht ein relativ langsames Zittern (Antagonistentremor – vier bis sechs Schläge pro Sekunde, selten bis neun Schläge pro Sekunde), das bei Bewegung abnimmt. Es ist typisch für das idiopathische Parkinson-Syndrom (75 %) und weniger typisch für atypische Parkinson-Syndrome (25 %), auch der Tremor ist einseitig betont. Der Tremor ist das augenfälligste Symptom, tritt aber auch als essentieller Tremor oder bei Kleinhirnerkrankungen usw. auf, so dass er zur Fehldiagnose verleiten kann.
Posturale Instabilität
Die verminderte Stabilität beim Aufrechthalten des Körpers kommt durch eine Störung der Stellreflexe zustande. Die kleinen, aber schnellen reflektorischen Ausgleichsbewegungen werden verzögert, so dass es zur Gang- und Standunsicherheit kommt. Die Wendebewegung wird unsicher, die Patienten kommen dabei ins Trippeln. Sie bekommen Angst zu fallen; diese Fallangst kann sie noch zusätzlich zur motorischen Behinderung beeinträchtigen. Bei früh stark gestörten Stellreflexen muss ein atypisches Parkinson-Syndrom bedacht werden.
Die unterschiedlichen Symptome können beim einzelnen Erkrankten unterschiedlich stark ausgeprägt sein oder ganz fehlen; Auftreten und Stärke wechseln auch im Tagesverlauf. Man unterscheidet daher die Verlaufsformen des Morbus Parkinson in „akinetisch-rigider Typ“, „tremordominanter Typ“ und „Äquivalenz-Typ“.
Fakultative Begleitsymptome
Neben diesen Kardinalsymptomen kommt es im Krankheitsverlauf in individuell unterschiedlichem Ausmaß zu weiteren Symptomen:
- Sensible Symptome
- eine Minderung des Geruchssinns (Hyposmie) ist häufig und kann der Parkinsonkrankheit oft bereits als initiales Symptom vorausgehen.
- Missempfindungen (Dysästhesien) werden häufig berichtet, ihre Ursache ist aber nicht genauer bekannt.
- Schmerzen treten besonders an Gelenken und Muskeln auf (siehe oben).
- Vegetative Störungen
- Ein Salbengesicht (fett-glänzende Gesichtshaut) entsteht durch gesteigerte Talgproduktion (zusammen mit der Hypomimie).
- Im fortgeschrittenen Krankheitsstadium kommt es zu Kreislaufregulationsstörungen (orthostatische Hypotonie). Nicht selten ist der Blutdruck im Liegen erhöht und sackt dann in aufrechter Körperhaltung ab, so dass die Patienten fälschlicherweise mit Medikamenten gegen hohen Blutdruck behandelt werden. Eine im Verlauf früh auftretende ausgeprägte Blutdruckinstabilität spricht für ein atypisches Parkinson-Syndrom.
- Blasenfunktionsstörungen behindern die Patienten im sozialen Leben erheblich. Meist steht zu Beginn ein plötzlicher starker Harndrang, oft schon bei kleinen Füllmengen (Pollakisurie). Das Auftreten von Miktionsstörungen früh im Verlauf (d. h. entweder vor oder innerhalb von drei Jahren nach Beginn motorischer Symptome) ist charakteristisch für ein atypisches Parkinson-Syndrom.
- Sexuelle Dysfunktionen sind häufig und betreffen sowohl die Libido als auch die Zeugungsfähigkeit.
- Bewegungsstörungen des Magen-Darm-Trakts können sowohl zu Durchfall als auch Verstopfung führen und die Resorption der Medikamente stark beeinflussen: Durchfall führt zu einer Unterdosierung, weil mehr von den verabreichten Wirkstoffen als pharmakologisch kalkuliert vorzeitig unresorbiert den Körper verlassen. Verstopfung führt zur Überdosierung, weil mehr von den verabreichten Wirkstoffen als pharmakologisch kalkuliert im Körper verbleiben und resorbiert werden; hierbei ergibt sich durch unterschiedliche Plasmahalbwertszeiten der Wirkstoffe zusätzlich eine unerwünschte Verschiebung ihrer Mengenverhältnisse.
- Temperatur-Regulationsstörungen führen vor allem zu einer verminderten Hitzetoleranz durch eine Störung des reflektorischen Schwitzens und der reflektorischen Gefäßerweiterung bei Wärme. Dies kann bei fortgeschrittener Erkrankung zu lebensbedrohlichen hochfieberhaften Zuständen führen. Besonders nachts kommt es zu starken Schweißausbrüchen.
- Im Verlauf frühzeitig auftretende vegetative Störungen weisen eher auf ein atypisches Parkinson-Syndrom.
- Psychische Veränderungen
- Eine niedergedrückte Stimmung kann als Frühsymptom der Diagnose um Jahre vorausgehen. Sie betrifft im Verlauf mindestens 40 Prozent der Patienten.
- Eine klassisch als Bradyphrenie bezeichnete Verlangsamung der Denkabläufe ist Ausdruck der allgemeinen Antriebsstörung. Sie gilt als Pseudodemenz, da das Denken nur verlangsamt, nicht aber inhaltlich beeinträchtigt ist.
- Die Störung der Einschätzung von Entfernungen und Geschwindigkeiten (Visuospatiale Aufmerksamkeit) stellt besonders in Verbindung mit den motorischen Einschränkungen eine Gefährdung im Straßenverkehr dar. Sie entspricht einer Störung im Frontalhirn.
- Sinnestäuschungen sind zumeist Folge der dopaminergen Medikamente. Sie führen zunächst zu benignen (= gutartigen) Halluzinationen, die der Betroffene als Trugbild erkennt. Zum Beispiel werden nicht vorhandene Personen im Raum gesehen. Dieses Symptom tritt erst im späteren Verlauf der Krankheit auf. Bei zusätzlicher subkortikaler Demenz können sich optische und akustische Halluzinationen weiter ausprägen bis hin zu einem meist als äußerst bedrohlich empfundenen szenischen Erleben, z. B. eingekerkert zu sein.[4][5] In diesem Zustand können die Patienten in panischer Angst aggressiv reagieren, was nicht selten verkannt wird und zu falschen therapeutischen Konsequenzen führt. Das den vor allem auch akustischen halluzinatorischen Sinnestäuschungen dauerhafte Ausgesetztsein kann sich verselbstständigen und zu nachhaltigen psychischen Erkrankungen wie Verfolgungswahn führen. Eine Besonderheit der kognitiven Störungen bei der Parkinson-Erkrankung ist die oft stark fluktuierende Störung der Aufmerksamkeit mit immer wieder luziden (klaren) Augenblicken.
- Eine echte Demenz stellt eine vermutliche Verlaufsform des idiopathischen Parkinson-Syndroms dar, die Lewy-Körperchen-Demenz.
- Die psychischen Veränderungen sind für die Alltagsbehinderung der Parkinson-Patienten von erheblicher Bedeutung und werden oft unterschätzt, da sie nicht so augenfällig sind wie die motorischen Phänomene.
- weitere Symptome
- REM-Schlafstörungen werden oft berichtet und können anderen Parkinson-Symptomen vorausgehen.
- Häufig existiert eine Assoziation mit Symptomen des Restless-Legs-Syndroms.
Diagnostik
Die Tatsache, dass die Symptomatik der Parkinson-Krankheit durch L-Dopa positiv beeinflussbar ist (siehe Abschnitt Behandlung), lässt sich diagnostisch nutzen. Beim sogenannten L-Dopa-Test wird die Schwere der Symptomatik mittels eines standardisierten Testes festgehalten (meist der motorische Teil III der Unified Parkinson’s Disease Rating Scale, UPDRS). Es folgt die Gabe einer definierten Menge an L-Dopa, meist das 1,5-fache der Vormedikation oder 100–200 mg L-Dopa plus ein Decarboxylasehemmer. Anschließend wird die Symptomatik erneut erfasst. Eine signifikante Verbesserung (z. B. > 30 % der UPDRS) der Symptomatik stützt, beweist jedoch nicht die klinische Diagnose eines idiopathischen Parkinson-Syndroms, sondern die Dopa-Sensitivität des Zielsymptomes.[3]
Ein bis zwei Tage vor Durchführung des L-Dopa-Tests wird die Gabe von Domperidon empfohlen, da L-Dopa bei zuvor unbehandelten Patienten zu deutlicher Übelkeit und Erbrechen führen kann.
Der Test kann auch mit dem Dopaminagonisten Apomorphin durchgeführt werden (Apomorphin-Test). Das Prinzip ist dasselbe, soll allerdings Dyskinesie-Symptome weniger wahrscheinlich machen. Er wird oft eingesetzt, wenn Patienten nicht primär L-Dopa erhalten sollen, wie dies bei jüngeren Patienten (hier: unter 70 Jahre) oft der Fall ist.
Zur Abgrenzung des Morbus Parkinson zu den Multisystematrophien kann die MIBG-Szintigrafie des Herzens eingesetzt werden.[6] Weitere nuklearmedizinische Methoden sind die Dopamin-Rezeptor-Szintigrafie und die Dopamin-Transporter-Szintigrafie.
Behandlung
Es gibt heute noch keine Möglichkeit einer ursächlichen Behandlung des Parkinson-Syndroms, die in einem Verhindern oder zumindest einem Aufhalten der fortschreitenden Degeneration der Nervenzellen des nigrostriatalen Systems bestünde. Daher muss man sich mit einer Behandlung der Symptome begnügen, die zunehmend gut möglich ist, was den Patienten, zumindest in den ersten Jahren (manchmal auch Jahrzehnten) der Erkrankung ein nahezu unbehindertes Leben ermöglicht.
Medikamentöse Behandlung
- Siehe auch: Parkinsonmittel
Die Behandlung erfolgt hauptsächlich durch die Gabe einer dopaminergen Medikation, das heißt, Medikamenten, die zu einer Erhöhung des Dopamin-Angebots im Gehirn führen bzw. Arzneistoffen, welche das fehlende Dopamin ersetzen.
Das wichtigste Medikament ist L-Dopa (Levodopa), eine Vorstufe des Dopamins. Dieser Vorstufe (Prodrug) ist es – im Gegensatz zum Dopamin selbst – möglich, die Blut-Hirn-Schranke zu durchqueren. Nach mehrjähriger Einnahme von L-Dopa können unwillkürliche Bewegungen, so genannte Dyskinesien, auftreten. Diese erklärt man durch eine pulsatile Rezeptorenstimulation, da L-Dopa nur eine Wirkzeit von wenigen Stunden hat. Deswegen empfiehlt man in der Regel, besonders bei jüngeren Patienten, zu Beginn der Parkinson-Krankheit die Behandlung mit einem länger wirkenden Dopaminagonisten. Dopaminagonisten ahmen an den Dopamin-Rezeptoren die Wirkung von Dopamin nach. Mit sogenannten MAO-B-Hemmern (Selegilin, Rasagilin) wird der Abbau von Dopamin im Gehirn verlangsamt.
Anticholinergika sollen dem relativen Überwiegen des Botenstoffs Acetylcholin gegenüber dem verminderten Dopamin entgegenwirken. Diese werden heute wegen ihres ungünstigen Nebenwirkungsprofils, insbesondere auf die kognitive Leistungsfähigkeit, nur noch selten verordnet. Sie spielen allerdings eine Rolle beim durch Neuroleptika induzierten sekundären Parkinson-Syndrom. Hemmstoffe der Catechol-O-Methyltransferase, so genannte COMT-Hemmer (Entacapon, Tolcapon), hemmen den Abbau der Dopaminvorstufe L-Dopa zu inaktiven Metaboliten. Dadurch erhöhen sie bei der gemeinsamen Einnahme mit Levodopapräparaten die Verfügbarkeit von Levodopa um 40 bis 90 Prozent und verlängern seine Plasmahalbwertszeit. Entacapon und Tolcapon dürfen nur in Verbindung mit L-Dopa und einem Decarboxylasehemmer angewendet werden. Diese Kombinationstherapie kann zu Einsparungen bei der Dosierung von Levodopa führen und somit das Nebenwirkungsprofil positiv beeinflussen. Ebenfalls eingesetzt wird Amantadin, besonders im Rahmen der akinetischen Krise.
Mit dem unaufhaltsamen Fortschreiten der Erkrankung muss die medikamentöse Behandlung im Verlauf immer wieder – durch einen Arzt für Neurologie oder in einer der Parkinson-Fachkliniken – angepasst werden.
Eine in Entwicklung befindliche Methode zur medikamentösen Einstellung im häuslichen Umfeld ist die Ambulante videounterstützte Parkinsontherapie.
L-Dopa-Präparate
L-Dopa-Präparate, von denen es in Deutschland mehr als 20 verschiedene gibt, enthalten immer L-Dopa in Kombination mit einem Decarboxylasehemmer (Carbidopa oder Benserazid), der den Abbau des gegen das Parkinson-Syndrom wirksamen L-Dopa peripher (das heißt im Organismus) hemmt, bevor es die Blut-Hirn-Schranke überwindet. So kommt man mit geringeren L-Dopa-Dosen aus und mit geringeren unerwünschten Wirkungen des Präparats außerhalb des Gehirns (wie zum Beispiel Herzrhythmusstörungen, Übelkeit, Mundtrockenheit).
Seit wenigen Jahren existiert ein Kombinationspräparat aus L-Dopa, Carbidopa und dem COMT-Hemmer Entacapon. Es ist bei Patienten angezeigt, bei denen zum Ende eines Dosisintervalls Fluktuationen auftreten, die mit einer Kombination aus L-Dopa mit nur einem Decarboxylasehemmer nicht ausreichend stabilisiert sind.
Dopaminagonisten
In der Therapie der Parkinson-Krankheit werden neben dem oben genannten Levodopa auch Arzneistoffe eingesetzt, die Dopamin-Rezeptoren stimulieren und somit eine dem Dopamin analoge Wirkung besitzen. Hierzu zählen die klassischen Mutterkornalkaloide (Bromocriptin, Cabergolin, Dihydroergocryptin, Lisurid und Pergolid) und die neueren selektiven D2-Rezeptoragonisten (Piribedil, Pramipexol, Ropinirol und Rotigotin). Die verschiedenen Präparate unterscheiden sich in ihrer Wirkdauer, im Wirkeintritt, in ihrer Galenik und in ihrem Nebenwirkungsprofil.
Eine weitere theoretische Behandlungsmethode stellt Amphetamin (ebenfalls ein Dopamin-Agonist) dar. Es führt zur erhöhten Ausschüttung von Dopamin in den synaptischen Spalt und hemmt dessen Wiederaufnahme in das präsynaptische Neuron.[7] Wegen der Nebenwirkungen und des Suchtpotentials sind Amphetamine derzeit keine zugelassenen Medikamente.
COMT-Hemmer
COMT-Hemmer sind Arzneistoffe, die das Dopamin und Levodopa abbauende Enzym Catechol-O-Methyl-Transferase kompetitiv hemmen. COMT-Hemmer werden immer in Kombination mit Levodopa eingesetzt. Durch die Hemmung des Abbaus und die Anreicherung von Levodopa in der Peripherie steigt die Aufnahme von Levodopa ins Zentralnervensystem und führt somit dort zu einer gewünschten Erhöhung der Dopaminkonzentration. Vertreter dieser Stoffgruppe sind Entacapon und Tolcapon. Letzteres war wegen schwerer, aber nur vereinzelt auftretender Leberschäden vorübergehend vom Markt genommen worden; es ist aber nach einer erneuten Sicherheitsbewertung von der europäischen Arzneimittelagentur (EMEA) für die Therapie der Parkinson-Krankheit wieder zugelassen worden, allerdings mit der Auflage der ständigen Kontrolle der Leberwerte.
Tiefe Hirnstimulation
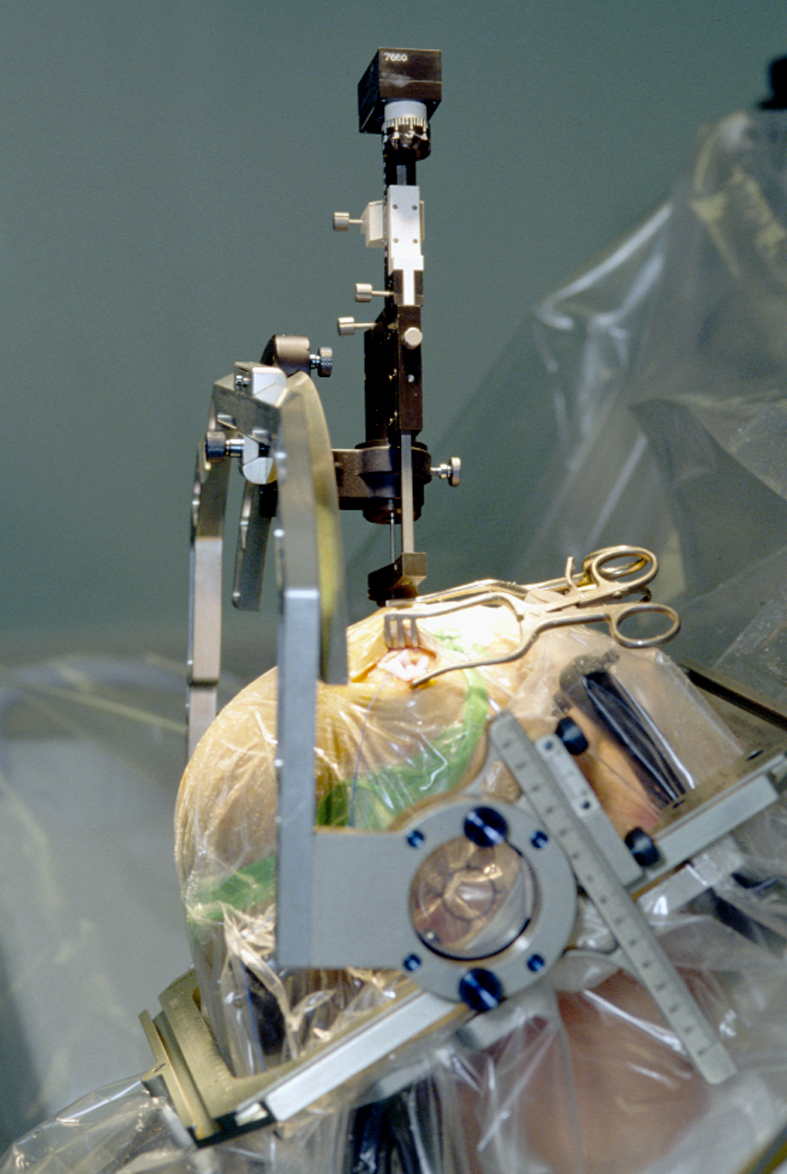
 Stereotaxiegerät zur Platzierung einer Stimulationselektrode
Stereotaxiegerät zur Platzierung einer StimulationselektrodeSeit vielen Jahren werden auch neurochirurgische Behandlungsmöglichkeiten eingesetzt. Eine seit Anfang der 1990er Jahre sehr erfolgreiche Methode ist die tiefe Hirnstimulation, bei der dem Patienten ein Impulsgenerator („Hirnschrittmacher“) eingesetzt wird.[8][9] Er erzeugt elektrische Impulse und leitet sie über dünne Kabel, je nach Lokalisation der Krankheitsursache und entsprechender Platzierung der Stimulationselektroden, in die jeweiligen Basalganglien Nucleus subthalamicus, Globus pallidus oder den vorderen Thalamus, wodurch dort überaktive Fehlimpulse wirksam unterdrückt werden können. Dieses Verfahren kommt bei Parkinson-Syndromen, daneben aber auch Dyskinesien und essentiellem Tremor in Frage, wenn die medikamentöse Therapie ihre Grenzen erreicht hat oder aus anderen Gründen nicht oder nur eingeschränkt einsetzbar ist.
Bei dem Eingriff zur Elektrodenplatzierung handelt es sich um eine schwierige und nicht ungefährliche stereotaktische Hirnoperation, die rund sechs bis zwölf Stunden dauert und sowohl im Vorfeld als auch während der Operation genaueste Planung und Kontrolle anhand von radiologisch gewonnenen räumlichen Bilddaten und elektrisch abgeleiteten neurophysiologischen Messwerten erfordert. Die Wirkung ist zumeist positiv. Es können jedoch eine vorübergehende oder länger andauernde Dysarthrie (Störung der Sprechmotorik) und ein meist auf ein Jahr begrenzter abnorm gesteigerter Antrieb auftreten. Durch die Verletzung der Hirnrinde können in seltenen Fällen auch Epilepsien ausgelöst werden. Ein entscheidender Vorteil der Methode gegenüber früheren „ablativen“ (zerstörenden) Verfahren, die bei diesen Krankheitsbildern heutzutage keine Anwendung mehr finden, liegt allerdings in der nahezu vollständigen Reversibilität.
Die Implantation des batteriebetriebenen Impulsgenerators selbst und dessen Kabelverbindung zur Stimulationselektrode wird in der Regel erst in einem zweiten chirurgischen Eingriff mehrere Tage nach der Elektrodenplatzierung vorgenommen. Das Gerät wird dabei, je nach seiner modellabhängigen Größe und der Physiognomie des betreffenden Patienten, in einer hierzu präparierten Hauttasche oberhalb des Brustmuskels oder im Bauchraum eingenäht. Nach dem zweiten Eingriff folgt eine Phase, in der die Stimulationseinstellungen des Impulsgebers individuell an die Symptome des Patienten angepasst und im Gerät programmiert werden. Sie kann durchaus mehrere Wochen in Anspruch nehmen und während dieser Zeit können auch noch vereinzelt oben genannte Nebenwirkungen (wie Dysarthrie, Dyskinesie, Dystonie) auftreten, weil sich das Gehirn des Patienten erst noch an die Stimulation von außen „gewöhnen“ muss.
Die Patienten erhalten anschließend ein spezielles Kontrollgerät, welches ihnen ermöglicht, selbstständig den Ladungszustand der Batterie regelmäßig zu überprüfen, den Stimulator bei Bedarf ein- und auszuschalten sowie bei einigen Modellen (nach vorheriger fachlicher Einweisung) auch die Stimulationseinstellungen des Impulsgebers innerhalb bestimmter voreingestellter Grenzbereiche selbst der jeweiligen Situation entsprechend anzupassen. Diese Kontrollgeräte arbeiten transkutan mittels magnetischen Impulsen. Je nach Einstellung des Impulsgenerators und ihrer Kapazität, halten die als Energiespeicher verwendeten Primärzellen („Batterie“) das Gerät etwa zwei bis fünf Jahre kontinuierlich in Betrieb. Sie sind fest im hermetisch gekapselten inerten Titan-Gehäuse des Gerätes eingebaut und somit praktisch untrennbar mit dem Gerät verbunden. Da sie sich deshalb weder einfach austauschen, noch bei den bisher verwendeten Geräten auch transkutan wieder aufladen lassen, ist nach entsprechenden Spannungsverlust durch die mehrjährige kontinuierliche Entladung die korrekte Funktion des Impulsgenerators nicht mehr gewährleistet. Zur Fortsetzung der Tiefen Hirnstimulation ist dann ein weiterer kurzer operativer Eingriff nötig, bei dem das alte Gerät entfernt und durch ein komplett neues ersetzt wird.
Forscher des Forschungszentrums Jülich und der Universität Köln arbeiten an der Entwicklung eines Hirnschrittmachers, der die Parkinson-Symptome nicht nur unterdrücken, sondern sie korrigieren und das Gehirn wieder normal funktionieren lassen soll.[10] Für diese Idee erhielten sie 2005 den Erwin-Schrödinger-Preis.
Implantation fetalen Hirngewebes
Die Implantation embryonaler Stammzellen in das Gehirn konnte zwar Ratten vom Parkinson-Tremor befreien, führte jedoch 2002 bei fünf von 19 Versuchstieren der Harvard Medical School zur Entwicklung unheilbarer Teratome. Der Forschungsansatz wird deshalb kaum noch weiterverfolgt. Vielversprechender sind therapeutische Ansätze mit weiterentwickelten Stammzellen. Dazu zählt der Einsatz neuraler Vorläuferzellen, die aus fetalem Gewebe isoliert worden sind. Sie sind von Wissenschaftlern des Rush-Presbyterian-St. Luke’s Medical Center in Chicago und des Universitätsklinikums Leipzig bereits an Ratten und Affen erfolgreich getestet worden und führten bei ihnen zu keinen negativen Nebenwirkungen. Eine in den USA durchgeführte doppelblind kontrollierte Pilot-Studie zur Wirksamkeit der Transplantation fetaler Vorläuferzellen der Substantia nigra zeigte keinen sicheren Effekt der Transplantationen (Annals of Neurology 2003, Olanow et al.). Neben den Komplikationen des Eingriffes selbst scheinen die Fluktuationen zwischen Unbeweglichkeit und Überbewegungen sogar noch zuzunehmen und unvorhersehbarer zu werden.
Komplementäre Behandlungsverfahren
Ausreichende Bewegung ist wichtig, um die für das Parkinson-Syndrom typische allmähliche Verminderung der Mobilität so lange wie möglich hinauszuzögern. Bei fortgeschrittener Krankheit ist dafür eine regelmäßige und speziell darauf ausgerichtete Physiotherapie nötig. Eine logopädische/sprachtherapeutische Unterstützung ist sinnvoll, wenn sich mit Fortschreiten der Erkrankung das Sprechen (leise und unexakte Aussprache, zu leise und zu hohe Stimme, zu schnelles Sprechen) und/oder das Schlucken (Verschlucken meist zunächst bei Flüssigkeiten, evtl. Komplikationen wie Lungenentzündungen) verschlechtert. Ergotherapie unterstützt durch Hilfen für den Alltag (z. B. Knöpfhilfen, Greifzangen) und arbeitet an der Raumwahrnehmung zur Verbesserung der Bewegung.
Alternativmedizinische Behandlungsmethoden
Etwa 40–60 % der Parkinson-Patienten nehmen – meist zusätzlich zur medikamentösen Therapie – alternativmedizinische Therapien in Anspruch. Unter diesen werden Entspannungs-, Meditations-, Atem- und Bewegungsübungen wie beispielsweise Taijiquan, Qigong, Yoga sowie Akupunktur und Massagen häufig angewendet. Einige Patienten verwenden Nahrungsergänzungsmittel wie beispielsweise Vitamine, um Mangelzuständen vorzubeugen oder vermeintliche Mangelzustände zu behandeln. Aussagekräftige klinische Studien, die eine Wirksamkeit dieser Behandlungen hinsichtlich der Lebensqualität und Symptomverbesserung untersucht haben, liegen nicht vor. Bei der Verwendung von Nahrungsergänzungsmitteln besteht die Möglichkeit schädlicher Wechselwirkungen mit der medikamentösen Therapie.[11] Vorsicht ist insbesondere bei der Einnahme von L-Dopa-haltigen Nahrungsergänzungsmitteln, zum Beispiel Extrakten der Juckbohne (Mucuna pruriens) geboten, da der L-Dopa-Gehalt hier oft schwankend ist und so zusammen mit einer medikamentösen L-Dopa-Therapie zu deutlichen Wirkschwankungen führen kann.
Selbsthilfegruppen
 Die Tulpe im Logo der Parkinson Selbsthilfe Österreich
Die Tulpe im Logo der Parkinson Selbsthilfe ÖsterreichAls eine der wertvollsten und beliebtesten Serviceeinrichtungen von Parkinson-Patientenorganisationen in aller Welt gilt die Entwicklung und Koordination eines Netzwerkes von Parkinson-Selbsthilfegruppen.
Eine Selbsthilfegruppe ist eine Gruppe aus lose zusammengesetzten Menschen, die sich alle in derselben Situation befinden und Erfahrungen und Probleme teilen. Gruppenteilnehmer unterstützen sich gegenseitig sowohl auf emotionaler als auch praktischer Ebene. Die Zusammenkunft mit anderen Betroffenen, die vor ähnlichen Herausforderungen stehen, bietet eine Gelegenheit, Gefühle, Kenntnisse und Erfahrungen auszutauschen. Vorteile einer aktiven Teilnahme sind einerseits praktische Tipps, aber auch eigene Motivation und die Inspiration, anderen zu helfen sich positiv mit den Veränderungen im Leben mit der Krankheit auseinanderzusetzen. Selbsthilfegruppen bieten darüber hinaus auch soziale Kontaktmöglichkeiten, die einer potentiellen Isolation entgegenwirken.
Selbsthilfegruppen treffen sich zu Gruppengesprächen, Vorträgen, gemeinsamen Freizeitunternehmungen und, seit dem Einzug des Internets, auch in Foren und virtuellen Räumen, wie zum Beispiel Parkins(on)line.[12]
Atypische Parkinson-Syndrome
Es gibt Krankheiten, die der Parkinsonschen Erkrankung ähneln und deren Ursache ebenfalls in einem Verfall von Nervenzellen im Bereich der Basalganglien im Gehirn liegt. Man nennt sie atypische Parkinson-Syndrome oder auch Parkinson-Plus-Syndrome: Menschen, die an diesen Krankheiten leiden, zeigen neben der Parkinson-Symptomatik weitere Symptome. Die häufigsten Krankheiten aus dem Bereich der atypischen Parkinson-Syndrome sind folgende:
- Multi-System-Atrophie (MSA)
- MSA-P Striatonigrale Degeneration (SND)
- MSA-C Olivopontocerebelläre Atrophie (OPCA)
- Primäre orthostatische Hypotension (Shy-Drager-Syndrom)
- Progressive supranukleäre Blickparese (PSP, auch: Steele-Richardson-Olszewski-Syndrom)
- Kortikobasale Degeneration (CBD)
Die atypischen Parkinson-Syndrome sind vergleichsweise selten. Allerdings gibt es eine hohe Dunkelziffer, eben weil diese Krankheitsbilder selten sind und die Patienten oft fehldiagnostiziert werden (als Morbus Parkinson, Morbus Alzheimer oder auch Depression). In Autopsien stellte sich z. B. die Lewy-Körperchen-Erkrankung (engl. Lewy body disease) als Ursache von ca. 50 Prozent der klinisch als „typisch“ diagnostizierten Parkinson-Syndrome heraus.
Siehe auch
Literatur
Monografien
- James Parkinson: Eine Abhandlung über die Schüttellähmung / An Essay on the Shaking Palsy, zweisprachige Ausgabe, Text von 1817, Neuausgabe Norderstedt 2009, ISBN 978-3-8370-2207-0.
- Reiner Thümler: Die Parkinson-Krankheit: Mehr wissen, besser verstehen. Trias, Stuttgart 2006, ISBN 3-8304-3321-2 (populärwissenschaftlich).
- Manfred Gerlach, Heinz Reichmann, Peter Riederer: Die Parkinson-Krankheit: Grundlagen, Klinik, Therapie. 3. Aufl., Springer, Wien/New York 2003, ISBN 3-211-83884-8.
- Gerd A. Fuchs: Die Parkinsonsche Krankheit: Ursachen und Behandlungsformen. C. H. Beck, München 2002, ISBN 3-406-48001-2.
- Reiner Thümler: Morbus Parkinson: Ein Leitfaden für Klinik und Praxis. Springer, Berlin/Heidelberg/New York/Barcelona/Hongkong/London/Mailand/Paris/Tokio 2002, ISBN 3-540-67471-3.
- Thomas Müller: Medikamentöse Therapie des Morbus Parkinson. UNI-MED-Verlag, Bremen /London /Boston 2005, ISBN 3-89599-864-8.
- Wolfgang H. Jost: Therapie des idiopathischen Parkinson-Syndroms. UNI-MED-Verlag, Bremen /London /Boston 2005, ISBN 3-89599-861-3.
- Siegfried Vogel, Reinhard Horowski: Leistung im Alter bei Parkinsonscher Krankheit: Ein Essay am Beispiel von Leonardo da Vinci, Wilhelm von Humboldt und Johannes Paul II. Duncker und Humblot, Berlin 2003, ISBN 3-428-11443-4.
- Susanne Schäfer: Die juvenilen und Young-onset-Parkinson-Syndrome. Verlag für Wissenschaft, Forschung und Technik, Wermelskirchen 2001, ISBN 3-929095-14-9.
Beschreibungen aus Patientensicht
- Michael J. Fox: Comeback: Parkinson wird nicht siegen. Bastei Lübbe, Bergisch Gladbach 2004, ISBN 3-404-61551-4 (Erfahrungsbericht).
- Wigand Lange: Wenn Parkinson kommt. Meine Erfahrungen mit einem ungebetenen Gast. Gütersloher Verlagshaus. 2007, 192 S., ISBN 3-579-06954-3
- Michael J. Fox: Lucky Man: A Memoir. Hyperion, 2002, 288 S., ISBN 0-7868-6764-7
- Helmut Dubiel: Tief im Hirn, Kunstmann, 2006, ISBN 3-88897-451-8
- Reinhard Hinterleitner: Mein Leben mit der Parkinsonkrankheit, 2001, ISBN 3-437-47400-6
Ratgeber für Angehörige
- Willibald Gerschlager (mit Hanne Brachtl, Wolfgang Freitag und Gerald Ganglbauer): Parkinson. 2009, 208 S., ISBN 978-3-85175-907-5
- Angelika Gollbach: Hilfe zur Selbsthilfe. Morbus Parkinson – Ratschläge (nicht nur) für Angehörige. 2007, 110 S., ISBN 978-3-926300-60-7
Zeitschriftenaufsätze
- Wolfgang Götz: Geschichte der Therapie des Morbus Parkinson – fast 200 Jahre keine kausale Therapie. Pharmazie in unserer Zeit 35 (3), S. 190–196 (2006), ISSN 0048-3664
- Hansruedi Büeler: Die Parkinson-Krankheit – Molekulare Mechanismen und Genetik. Pharmazie in unserer Zeit 35 (3), S. 198–203 (2006), ISSN 0048-3664
- Bernd Riebesehl, Ralph Lipp: Darreichungsformen in der Parkinson-Therapie – Arzneiformen eröffnen neue Wege für Parkinson-Patienten. Pharmazie in unserer Zeit 35 (3), S. 226–233 (2006), ISSN 0048-3664
- Jan Schindehütte, Walter Paulus, Ahmed Mansouri: Stammzellentherapie bei Morbus Parkinson – Zellersatz als eine therapeutische Option? Pharmazie in unserer Zeit 35 (3), S. 250–254 (2006), ISSN 0048-3664
- Jowaed A., Schmitt I., Kaut O., Wüllner U.: Methylation regulates alpha-synuclein expression and is decreased in Parkinson's disease patients' brains. In: J Neurosci. 30 (18), Nr. 5, 2010, S. 6355-9, PMID 20445061.
Sonstige Werke (Belletristik, Drama etc.)
- Klaus Fehling: Nicht mein Bein. Hörspiel. Produktion: WDR 2007, ISBN 978-3-942792-04-2
- Markus Dietrich: Mein Robodad. Kurzfilm. Prod: Gruppe Weimar
Weblinks
Übersicht
- Parkinson-Krankheit: Epidemiologie, Diagnose, Verlauf, Ätiologie, Komorbidität, Behandlung, Prävention, Literatur
- S2-Leitlinie Parkinson-Syndrome, Diagnostik und Therapie der Deutschen Gesellschaft für Neurologie. In: AWMF online (Stand 2008)
- www.kompetenznetz-parkinson.de Glossar (= Erklärung relevanter Fachbegriffe)
Forschung
- Deutsche Parkinson Gesellschaft
- Kompetenznetz Parkinson
- Michael J. Fox Stiftung für Parkinsonforschung (englisch)
- Parkinson-Forschung in den USA (englisch, spanisch)
- Artikelsammlung zur Parkinson-Stammzellenforschung (en)
Patientenorganisationen
Einzelnachweise
- ↑ American Academy of Neurology vom 20. Februar 2011: Using Amphetamines May Increase Risk of Parkinson’s Disease (eingesehen am 23. Februar 2011)
- ↑ James Parkinson: An Essay on the Shaking Palsy. Sherwood, Neely, and Jones, London 1817 (Onlinescan der Originalausgabe).
- ↑ a b Leitlinie Parkinson-Syndrome der DGN als PDF
- ↑ Freedman M. Parkinson's disease. In: Cummings JL, ed. Subcortical dementia. New York: Oxford University Press, 1990: 108-122.
- ↑ Beatty WW, Ryder KA, Gontkovsky ST, Scott JG, McSwan KL, Bharucha KJ. Analyzing the subcortical dementia syndrome of Parkinson's disease using the RBANS. Arch Clin Neuropsychol 2003;18(5): S. 509-520.
- ↑ S. Braune, M. Reinhardt, R. Schnitzer, A. Riedel, C. H. Lücking: Cardiac uptake of [123I]MIBG separates Parkinson's disease from multiple system atrophy. In: Neurology. 53, Nr. 5, September 1999, S. 1020–1025. PMID 10496261.
- ↑ Silbernagl, Stefan; Lang, Florian: Taschenatlas der Pathophysiologie. Thieme Verlag 1998, Stuttgart, New York. S. 312 f.
- ↑ Charité – Klinik für Neurologie, AG Bewegungsstörungen: „Hirnschrittmacher“ gegen die Parkinson-Erkrankung – Eine Patientenaufklärung (Kupsch, A.; Ulm, G.; Funk, T.) [PDF]
- ↑ dPV e. V. – „Club U40 im Internet“: Hirnschrittmacher gegen die Parkinson-Erkrankung (Kupsch, A.; Ulm, G.; Funk, T.)
- ↑ Handelsblatt: Gezielt aus dem Takt gebracht
- ↑ M. J. Nijkrake, S. H. J. Keus, J. G. Kalf et al.: Allied health care interventions and complementary therapies in Parkinson’s disease. In: Parkinsonism Relat Disord. 2007;13 Suppl 3: S. 488–494. PMID 18267288
- ↑ Parkins(on)line, Website der virtuellen Parkinson-Selbsthilfe, abgerufen am 1. Juli 2011.

Bitte den Hinweis zu Gesundheitsthemen beachten!

Wikimedia Foundation.