- Langmuir - Hinshelwood - Mechanismus
-
Als Katalyse (griechisch κατάλυσις, katálysis – die Auflösung, Abschaffung, Aufhebung; ursprünglich von: κατά = „(da)bei“ (örtl./zeitl.) und λύσις = „Auflösung, Zerfall“, also: „örtlich und zeitlich beim Zerfall (anwesend)“, wörtlich: „der Bei-Zerfall“) wird die Veränderung der Reaktionsgeschwindigkeit einer chemischen Reaktion durch Beteiligung eines Katalysators ohne die Veränderung des thermodynamischen Gleichgewichts bezeichnet.
Der Katalysator geht unverändert aus der Gesamtreaktion wieder hervor und kann somit mehrere Katalysezyklen durchlaufen, während der Substrat genannte Ausgangsstoff abgebaut und das Produkt aufgebaut wird.
Da über 80 % aller Chemieerzeugnisse während der Herstellung mit Katalysatoren in Berührung kommen, ist die Wertschöpfung durch diese sehr hoch und von erheblicher volkswirtschaftlicher Bedeutung.
Geschichte der Katalyse
Als erste bekannte katalytische Prozesse gelten die Alkoholvergärung aus Zucker sowie die Essigsäureherstellung aus Alkohol mit Hilfe von katalytisch wirkenden Enzymen.
Im 17ten und frühen 18ten Jahrhundert wurden eine ganze Reihe von neuen katalytischen Reaktionen gefunden. Antoine-Augustin Parmentier entdeckte 1781 die Stärkespaltung zu Zucker unter Säurekatalyse, Carl Wilhelm Scheele entdeckte 1782 die säurekatalysierte Veresterung von Alkoholen und Säure zu Estern und Joseph Priestley 1783 den Zerfall von Ethanol zu Ethylen und Wasser an Tonerde.
Jahr Reaktion Katalysator Entdecker 1781 Zerfall von Stärke in Traubenzucker H+ Parmentier 1782 Veresterung H+ Scheele 1806 Bleikammerverfahren NO2 Desormes, Clement[1] 1808 Ammoniakzerfall zu Stickstoff und Wasserstoff Fe Berthollet 1818 Zerfall von Wasserstoffperoxid Ag, Ag2O, MnO2 Thénard 1823 Entzündung von Wasserstoff (Döbereiners Feuerzeug) Pt Döbereiner  Wilhelm Ostwald
Wilhelm Ostwald1835 erkannte Jöns Jakob Berzelius in den obigen Reaktionen die Gemeinsamkeit, dass neben den Edukten und Produkten immer ein weiterer Stoff in der Reaktion notwendig war, der aber anscheinend nicht verbraucht wurde. Er prägte dazu den Begriff Katalyse in Analogie zu Analyse.[2]
Eine moderne Definition der Katalyse fand aber erst 1894 Wilhelm Ostwald. Diese lautet: "Katalyse ist die Beschleunigung eines langsam verlaufenden chemischen Vorgangs durch die Gegenwart eines fremden Stoffes"[3]. Später spezifiziert zu: „Ein Katalysator ist ein Stoff, der die Geschwindigkeit einer chemischen Reaktion erhöht, ohne selbst dabei verbraucht zu werden und ohne die endgültige Lage des thermodynamischen Gleichgewichts dieser Reaktion zu verändern.“ Als Anerkennung für seine Arbeiten über die Katalyse sowie für seine grundlegenden Untersuchungen über chemische Gleichgewichtsverhältnisse und Reaktionsgeschwindigkeiten wurde Ostwald im Jahre 1909 mit dem Nobelpreis für Chemie ausgezeichnet.
Energetische Grundlagen der Katalyse
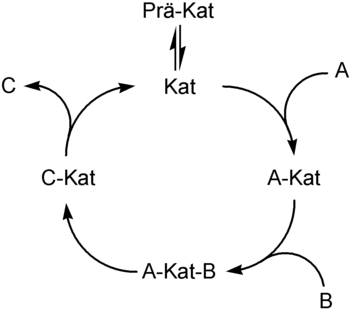
 Energiediagramm einer enzymatischen Reaktion: Die Aktivierungsenergie (freie Aktivierungsenthalpie) wird im Vergleich zu unkatalysierten Reaktionen durch Stabilisierung des Übergangszustandes gesenkt. Die freie Reaktionsenthalpie dagegen bleibt unverändert.
Energiediagramm einer enzymatischen Reaktion: Die Aktivierungsenergie (freie Aktivierungsenthalpie) wird im Vergleich zu unkatalysierten Reaktionen durch Stabilisierung des Übergangszustandes gesenkt. Die freie Reaktionsenthalpie dagegen bleibt unverändert.Eine einfache chemische Reaktion A + B → AB kann z. B. folgendermaßen durch einen Katalysator Kat. beeinflusst werden, wobei AKat. dem Übergangszustand entspricht:
- A + Kat. → AKat.
- AKat. + B → AB + Kat.
Katalysatoren erhöhen die Reaktionsgeschwindigkeit von chemischen Reaktionen um mehrere Größenordnungen. Es ist allerdings nicht möglich, Reaktionen durchzuführen, die thermodynamisch verboten sind in der Hinsicht, dass die Gesamtenergie der Produkte höher liegt als die der Ausgangsstoffe, das heißt, dass die Katalyse eine kinetische und keine thermodynamische Erscheinung ist. Wie bei jeder spontan ablaufenden Reaktion muss die freie Reaktionsenthalpie (ΔG) negativ sein. Das chemische Gleichgewicht wird durch einen Katalysator nicht verändert, wohl aber die Geschwindigkeit, mit der es sich einstellt. Die katalytische Wirksamkeit beruht einzig auf der Fähigkeit, die Aktivierungsenergie
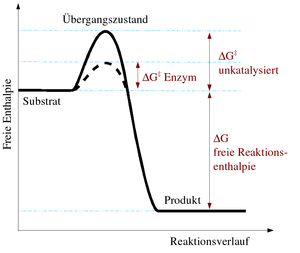
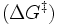 einer chemischen Reaktion zu senken. Die Aktivierungsenergie ist der Energiebetrag, der zunächst überwunden werden muss, um die Reaktion in Gang zu setzen. Während der Reaktion wird das Substrat zunehmend verändert, es nimmt einen energetisch ungünstigen Übergangszustand ein. Die Aktivierungsenergie ist nun der Energiebetrag, der benötigt wird, um das Substrat in den Übergangszustand zu zwingen. Hier setzt die Wirkung des Katalysators ein, da er durch Wechselwirkung mit dem Übergangszustand diesen stabilisiert und daher weniger Energie benötigt wird, um das Substrat in den Übergangszustand zu bringen. Aus der Arrhenius-Gleichung ergibt sich der Effekt einer niedrigen Aktivierungsenergie bei sonst gleichen Reaktionsbedingungen auf die Reaktionsgeschwindigkeitskonstante:
einer chemischen Reaktion zu senken. Die Aktivierungsenergie ist der Energiebetrag, der zunächst überwunden werden muss, um die Reaktion in Gang zu setzen. Während der Reaktion wird das Substrat zunehmend verändert, es nimmt einen energetisch ungünstigen Übergangszustand ein. Die Aktivierungsenergie ist nun der Energiebetrag, der benötigt wird, um das Substrat in den Übergangszustand zu zwingen. Hier setzt die Wirkung des Katalysators ein, da er durch Wechselwirkung mit dem Übergangszustand diesen stabilisiert und daher weniger Energie benötigt wird, um das Substrat in den Übergangszustand zu bringen. Aus der Arrhenius-Gleichung ergibt sich der Effekt einer niedrigen Aktivierungsenergie bei sonst gleichen Reaktionsbedingungen auf die Reaktionsgeschwindigkeitskonstante:mit A präexponentieller Faktor oder Frequenzfaktor, entspricht nach der Stoßtheorie dem Produkt aus der Stoßzahl Z und dem Orientierungsfaktor P,
EA Aktivierungsenergie (Einheit: J/mol),
R = 8,314 J/(K mol) allgemeine Gaskonstante,
T absolute (thermodynamische) Temperatur (Einheit: K).
k ReaktionsgeschwindigkeitskonstanteKatalysator
Ein Katalysator ist ein Stoff, der die Reaktionsgeschwindigkeit einer chemischen Reaktion verändert, ohne dabei verbraucht zu werden. Der Katalysator beschleunigt eine Reaktion bzw. setzt die notwendige Aktivierungsenergie herab, so dass eine Reaktion bei niedrigeren Temperaturen stattfinden kann. Viele Reaktionen sind überhaupt erst durch einen Katalysator möglich, weil die Reaktion ansonsten extrem langsam ablaufen würde. Siehe auch:
- Fahrzeugkatalysator
- Grubbs-Katalysatoren
- Hopcalite
- Lindlar-Katalysator
- Raney-Nickel
- Wechselzahl
- Wilkinson-Katalysator
- Ziegler-Natta-Katalysator
Co-Katalysator
Oft benötigt ein katalytischer Zyklus neben dem eigentlichen Katalysator einen Co-Katalysator, der die Aktivität oder Selektivität eines Katalysators in positiver Weise beeinflusst. Ein bekanntes Beispiel ist die Funktion von Methylaluminoxan als Co-Katalysator in der Olefin-Polymerisation sowie die Jodwasserstoffsäure im Monsanto-Prozess.
Einteilung der Katalyse
Je nachdem, ob in welcher Phase Katalysator und Substrat vorliegen, wird nach homogener (Katalysator und Substrat liegen in der gleichen Phase vor) und heterogener Katalyse (Katalysator und Substrat liegen in verschiedenen Phasen vor) unterschieden. Wechselt der Katalysator oder das Substrat während der Reaktion die Phase, spricht man auch von Phasentransferkatalyse.
Bei der homogenen Katalyse können der Katalysator und das Substrat entweder in der Gasphase, etwa NO2 und SO2 beim Kontaktverfahren, oder in der flüssigen Phase vorliegen.
Bei der heterogenen Katalyse liegt der Katalysator oft in fester Form vor (sogenannte Kontakte) und das Substrat reagiert aus der Gas- oder Flüssigphase.
Je nach Art des verwendeten Katalysators gibt es weitere Einteilungskriterien wie Übergangsmetallkatalyse, Säure-Base-Katalyse oder Biokatalyse. Weisen die Produkte einer katalytischen Reaktion spezielle stereochemische Eigenschaften auf, so spricht man z. B. von enantioselektiver Katalyse. Wirkt ein während einer Reaktion hergestellter Stoff katalytisch auf die Erzeugungsreaktion, so nennt man diesen Vorgang Autokatalyse.
Werden Reaktionen mit Hilfe organischer Moleküle katalysiert, spricht man von Organokatalyse. Der Katalysator enthält in der Regel kein Metall und besteht nur aus leichten Hauptgruppenelementen.[4]
Mechanismen in der homogenen Katalyse
Säure-Base-Katalyse
Ein bekanntes Beispiel einer säurekatalysierten Reaktion ist die bereits oben erwähnte Veresterung von Carbonsäuren mit Alkoholen bzw. die Umkehrung dieser Reaktion unter Verseifung (für die Reaktionsmechanismen siehe Artikel Veresterung, Verseifung).
Übergangsmetallkatalyse
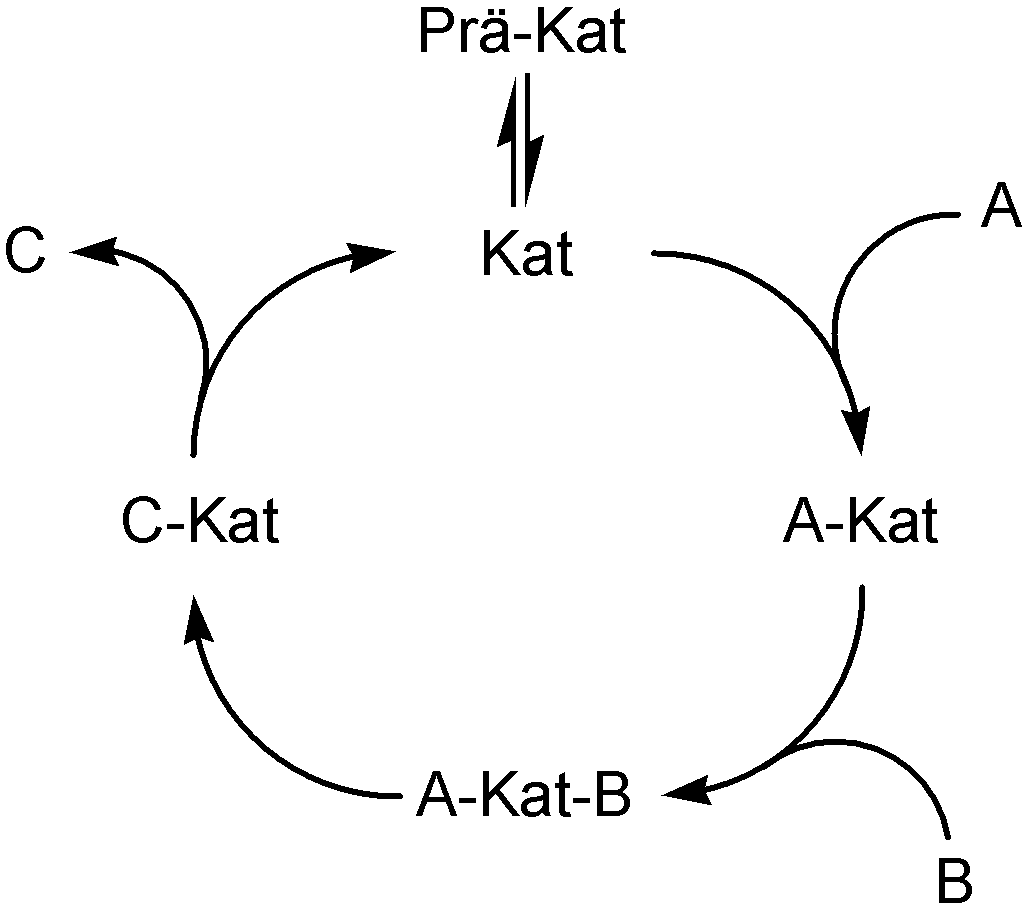
Die Übergangsmetallkatalyse beinhaltet oft die Schritte
- Bildung der aktiven Katalysatorspezies (Prä-Kat -> Kat)
- oxidative Addition eines Substrats an den Katalysator (A + Kat -> A-Kat)
- Komplexbildung eines Substrats mit dem Katalysator (A-Kat + B -> A-Kat-B)
- Insertion eines Substrats in eine Substrat-Katalysator-Bindung (A-Kat-B -> C-Kat)
- gegebenenfalls eine Umlagerung
- reduktive Eliminierung des Produktes unter Freisetzung des Katalysators und des Produkts (C-Kat -> C + Kat)
die einen Katalysezyclus ergeben.
Siehe auch:
Biokatalyse
Stoffwechselvorgänge in Lebewesen werden durch Enzyme katalysiert. Diese Reaktionen zeichnen sich allgemein durch äußerst hohe Effizienz und Selektivität aus und laufen bei milden Temperaturen und in wässrigem Milieu ab. Reaktive Spezies, die mit Wasser reagieren würden, werden durch hydrophobe „Taschen“ abgeschirmt. Viele Biokatalysatoren sind Proteine oder enthalten Proteinbestandteile.
Siehe auch:
Mechanismen in der heterogenen Katalyse
Bei monomolekularen Reaktionen zerfällt z. B. einfach das Edukt an der Katalysatoroberfläche. Bei bimolekularen Reaktionen sind drei Mechanismen denkbar:
Langmuir-Hinshelwood-Mechanismus
Beim Langmuir-Hinshelwood-Mechanismus werden zunächst beide Edukte A und B aus der Gasphase auf der Katalysatoroberfläche adsorbiert:
- Ag → Aads
- Bg → Bads
Anschließend reagieren die adsorbierten Edukte auf der Katalysatoroberfläche zu Produkt C ab:
- Aads + Bads → Cads
Im letzten Schritt desorbiert Produkt C:
- Cads → Cg
Eley-Rideal-Mechanismus
Beim Eley-Rideal-Mechanismus adsorbiert zunächst Edukt A auf der Katalysatoroberfläche:
- Ag → Aads
Anschließend reagiert das adsorbierte Edukt mit einem weiteren Edukt B aus der Gasphase zum Produkt C:
- Aads + Bg → Cads
Im letzten Schritt desorbiert Produkt C:
- Cads → Cg
Mars-van-Krevelen-Mechanismus
Edukt A wird zunächst aus der Gasphase auf der Katalysatoroberfläche adsorbiert:
- Ag → Aads
Anschließend folgt die Oxidation von Edukt A mit vorhandenem Gittersauerstoff:
- Aads + Osurf → AOads
Produkt AO desorbiert und es entsteht eine Sauerstoffleerstelle im Kristallgitter:
- AOads → AOg + Leerstelle
Nach der Desorption werden durch Reoxidation mit Sauerstoff die Leerstellen wieder aufgefüllt:
- O2,g → 2 Oads → 2 Osurf
Trägermaterialien
 Honeycomb aus Cordierit
Honeycomb aus CordieritTrägermaterialien tragen in der heterogenen Katalyse die feinverteilten, katalytisch wirksame Metallcluster und können aufgrund ihrer Eigenschaften auch als Co-Katalysator dienen, oder sie haben als Ligand einen Einfluss auf die katalytische Aktivität des dispergierten Metalls. Durch besondere Strukturen, z. B. bei Zeolithen, können Trägermaterialien auch die Selektivität einer Reaktion beeinflussen. Beispiele von Trägermaterialien sind:
- Cordierit
- Russ
- Silicagel
- Zeolithe
- Metalloxide, z. B. Titandioxid, Aluminiumoxid
Siehe auch:
Phasentransferkatalyse
Vermittelt ein Katalysator den Kontakt zweier Reaktanden, die in unterschiedlichen Phasen (meist wässrige und organische Phase) vorliegen, so spricht man von Phasentransferkatalyse. Der Katalysator einer solchen Reaktion ermöglicht den Durchtritt der Reaktanden durch die Phasengrenze.
Beispielsweise ermöglichen Kronenether die Lösung von Alkalimetallionen in organischen Lösungsmitteln; die Anionen, z. B. MnO4-, werden dann als Kronenether/Alkali-Anion Ionenpaar in die organische Phase geschleppt. Dabei haben sich Kronenether mit 15 und 18 Kohlenstoffatomen im Ring besonders bewährt. Auch quartäre Ammoniumverbindungen, Phosphonium- oder Arsoniumionen mit lipophilen Alkylresten verbessern die Extraktion. Dabei steigt die Lipophilie mit zunehmendem Kohlenstoffanteil im Alkylrest. Die Reaktionsgeschwindigkeit einer Phasentransferkatalyse steigt oft linear mit der Konzentration des Katalysators an.
Katalysatordeaktivierung
Die Mechanismen der Katalysatordeaktivierung sind vielfältig. In der Übergangsmetallkatalyse wird die Reduktion der eingesetzten Metall-Ligand-Katalysatoren zum Metall beobachtet, in der heterogenen Katalyse spielt bei Raffinerieprozessen die Verkokung oder Sinterung der aktiven Oberfläche eine große Rolle. Siehe auch:
Bekannte Forscher auf dem Gebiet der Katalyse
Die Forschungsgebiete auf dem Gebiet der Katalyse sind vielfältig. In der homogenen Katalyse wird z. B. der Einfluss der Liganden, des Lösungsmittels sowie die Trennung von Katalysator und Produkt untersucht. In der heterogenen Katalyse stehen die Untersuchungen der chemischen Prozesse and Oberflächen im Vordergrund, für die Ertl mit dem Nobelpreis für Chemie im Jahr 2007 ausgezeichnet wurde, aber auch die Entwicklunge neuer Trägermaterialien. Für Forschungen im Bereich Katalyse werden viele wissenschaftliche Preise vergeben, z. B. der Jochen-Block-Preis der DECHEMA. Siehe auch:
- Manfred Baerns
- Jöns Jakob Berzelius
- Georg Bredig
- Johann Wolfgang Döbereiner
- Gerhard Ertl
- Henri Kagan
- Helmut Knözinger
- Irving Langmuir
- Paul Sabatier
Bekannte Institute und Forschungseinrichtungen
- Department für Chemie der Technischen Universität München
- Leibniz-Institut für Katalyse
- Max-Planck-Institut für Kohlenforschung
Beispiele für Katalytische-Prozesse
- die Reduktion von Stickoxiden bei gleichzeitiger Oxidation von Kohlenmonoxid und Kohlenwasserstoffen aus den Motorabgasen im Dreiwege-Katalysator von Kraftfahrzeugen
- die Oxidation von Wasserstoff und Sauerstoff zu Wasser in Brennstoffzellen
- nahezu alle biochemischen Vorgänge werden durch Enzyme katalysiert.
Reaktion Katalysator Verfahren Entdecker Jahr H2SO4 aus SO2, H2O und Luft NO2 Bleikammerverfahren Desormes, Clement 1806 Ammoniak aus N2 und H2 Fe Haber-Bosch-Verfahren Haber, Bosch, Mittasch 1910 HNO3 aus NH3 und Luft Pt/Rh Ostwald-Verfahren Wilhelm Ostwald Aldehyde aus Olefinen und O2 Pd Wacker-Verfahren 1913 Alkane und Olefine aus Alkanen Al, Si Fluidized-Bed-Catalytic-Cracken Alkane aus Alkanen Ni, Al, Si Hydrocracken Alkane aus CO/H2 Fe, Co Fischer-Tropsch-Verfahren Fischer, Tropsch 1925 Aldehyde aus CO und H2 Rh, Co Hydroformylierung Otto Roelen 1938 Aromaten, H2, Isoalkane aus n-Alkanen Pt, Sn, Re, Al, Si Katalytisches Reforming Vladimir Haensel 1948 Nitrile aus HCN und Olefinen Ni Hydrocyanierung Polyethylen aus Ethylen Ti/Al Niederdruckverfahren Karl Ziegler 1953 Cyclooctadien aus Butadien Ni Günther Wilke α-Olefine aus Ethen Ni SHOP-Prozess Wilhelm Keim Olefine aus Olefinen Ru Metathese u. a. Chauvin, Schrock, Grubbs Essigsäure aus Methanol und CO Rh, Ir Monsanto-Prozess Foster Einzelnachweise
- ↑ Grundlagen der metallorganischen Komplexkatalyse von Dirk Steinborn; Google-Books
- ↑ G. Ertl, T. Gloyna, Z. Phys. Chem. 2003, 217(10), 1207–1219.
- ↑ W. Ostwald, "Referat zur Arbeit F. Strohmann Über den Wärmegehalt der Bestandteile der Nahrungsmittel" Z. phys. Chem. 15 (1894), S. 705 f.
- ↑ Skript der Universität Hannover über Organokatalyse
Literatur
Ältere Literatur
- Walther Gerlach: Julius Robert Mayer. Die Chemie (Angewandte Chemie, neue Folge) 55(49/50), S. 369 – 375 (1942), ISSN 1521-3757
- A. Mittasch: Wesentliches und Abseitiges zur Geschichte der „katalytischen Kraft“. Die Chemie (Angewandte Chemie, neue Folge) 55(49/50), S. 375 – 376 (1942), ISSN 1521-3757
Aktuelle Literatur
- P. Kripylo, K.-P. Wendlandt, F. Vogt: Heterogene Katalyse in der chemischen Technik. Deutscher Verlag fur Grundstoffindustrie GmbH, Leipzig, 1993
- G. Ertl: Activation of diatomic molecules at solid surfaces. Phil. Trans. R. Soc. A 363, 955-958, 2005
- Gerhard Ertl: Reaktionen an Oberflächen: vom Atomaren zum Komplexen (Nobel-Vortrag) (engl.)
- Gabor A. Somorjai: Introduction to Surface Chemistry and Catalysis. Wiley, New York 1994, ISBN 0-471-03192-5 (Englisch).
Siehe auch
- Biokatalysator
- Katalysator
- Katalysatoraktivität
- Katalysatorwirkungsgrad
- Organokatalyse
- Photokatalyse
Weblinks

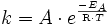

Wikimedia Foundation.