- Thermodynamik
-
Die Thermodynamik (von altgriechisch θερμός thermós „warm“ sowie δύναμις dýnamis „Kraft“),[1] auch als Wärmelehre bezeichnet, ist ein Teilgebiet der klassischen Physik. Sie beschäftigt sich mit der Möglichkeit, durch Umverteilen von Energie zwischen ihren verschiedenen Erscheinungsformen Arbeit zu verrichten. Die Grundlagen der Thermodynamik wurden aus dem Studium der Volumen-, Druck-, Temperaturverhältnisse bei Dampfmaschinen entwickelt.
Man unterscheidet zwischen offenen, geschlossenen und abgeschlossenen (isolierten) thermodynamischen Systemen. Bei einem offenen System fließt – im Gegensatz zum geschlossenen – Materie über die Systemgrenze, abgeschlossene Systeme sind auch energiedicht. Nach dem Energieerhaltungssatz bleibt darin die Summe aller Energieformen (thermische, chemische, Federspannung, Magnetisierung usw.) konstant.
Die Thermodynamik bringt die Prozessgrößen Wärme und Arbeit an der Systemgrenze mit den Zustandsgrößen in Zusammenhang, welche den Zustand des Systems beschreiben. Dabei wird zwischen intensiven Zustandsgrößen (beispielsweise Temperatur T, Druck p, Konzentration n und chemisches Potential μ) und extensiven Zustandsgrößen (beispielsweise innerer Energie U, Entropie S, Volumen V und Teilchenzahl N) unterschieden.
Auf der Basis von vier fundamentalen Hauptsätzen sowie materialspezifischen, empirischen Zustandsgleichungen zwischen den Zustandsgrößen (s. z. B. Gasgesetz) erlaubt die Thermodynamik durch die Aufstellung von Gleichgewichtsbedingungen Aussagen darüber, welche Änderungen an einem System möglich sind (beispielsweise welche chemischen Reaktionen oder Phasenübergänge ablaufen können, aber nicht wie) und welche Werte der intensiven Zustandsgrößen dafür erforderlich sind. Sie dient zur Berechnung von frei werdender Wärmeenergie, von Druck-, Temperatur- oder Volumenänderungen, und hat daher große Bedeutung für das Verständnis und die Planung von Prozessen in Chemieanlagen, bei Wärmekraftmaschinen sowie in der Heizungs- und Klimatechnik.
Die Thermodynamik macht aber keine Aussagen darüber, wie schnell die Prozesse ablaufen (Kinetik), sodass es Bestrebungen gab, den Begriff Thermodynamik durch Thermostatik zu ersetzen.[2]
Durch die statistische Mechanik nach James Clerk Maxwell und Ludwig Boltzmann können viele Aspekte der Thermodynamik anhand mikroskopischer Theorien bestätigt werden. In ihrer gesamten Darstellung behält sie allerdings weiterhin den ausgezeichneten Status einer eigenständigen physikalischen Theorie. Ihre Anwendbarkeit muss jedoch auf geeignete Systeme eingeschränkt werden, nämlich solche, die sich aus genügend vielen Einzelsystemen, also meist Teilchen, zusammensetzen.
 Typischer thermodynamischer Vorgang am Beispiel der prinzipiellen Wirkungsweise eines durch Dampf betriebenen Motors (rot = sehr heiß, gelb = weniger heiß, blau = Endtemperatur des Mediums)
Typischer thermodynamischer Vorgang am Beispiel der prinzipiellen Wirkungsweise eines durch Dampf betriebenen Motors (rot = sehr heiß, gelb = weniger heiß, blau = Endtemperatur des Mediums)
Geschichte
Der französische Physiker Nicolas Léonard Sadi Carnot untersuchte die Wärmemengen einer Dampfmaschine (1824). Er stellte fest, dass heißer Wasserdampf ein kälteres Wasserreservoir erwärmt und dabei mechanische Arbeit geleistet wird. Carnot vermutete, dass bei diesem Prozess keine Wärme verloren geht. Carnot beschrieb die Vorgänge in der Dampfmaschine als Kreisprozess, die in späteren Jahren von Benoît Pierre Émile Clapeyron in mathematischer Form dargestellt wurde (Carnotscher Kreisprozess).[3]
Der deutsche Arzt Julius Robert Mayer formulierte (1841) die These, dass Energie in einem abgeschlossenen System eine konstante Größe sein sollte. Energie kann nicht verschwinden, sondern nur in eine andere Form umgewandelt werden. Diese Erkenntnis ist als Energieerhaltungssatz bekannt. Mayer machte Berechnungen zur Umwandlung von Wärme in mechanische Energie. Er gab an, wie viel Energie die Temperaturerhöhung von 1 g Wasser um 1 °C entspricht und berechnete, dass diese Energiemenge einer mechanischen Energie entspricht, die 1 g Materie 367 Meter in die Höhe heben könnte (tatsächlich sind es 426 Meter). Diese Berechnungen bildeten die Grundlage zum Ersten Hauptsatz der Thermodynamik.[4] James Prescott Joule bestimmte im Jahr 1844 noch genauer das mechanische Wärmeäquivalent.
Im Jahr 1840 veröffentlichte der deutsch-schweizerische Chemiker Hermann Heinrich Hess eine Abhandlung mit dem Titel „Thermochemische Untersuchungen“, die auf dem Satz von der Erhaltung der Energie bei Molekülen bzw. Atomen aufgrund von chemischen Reaktionswärmen basierte.
Während Carnot noch vermutete, dass die Wärmemengen bei einer Dampfmaschine vollständig erhalten bleiben, nahm Mayer eine Umwandelbarkeit von Energieformen ineinander an. Der deutsche Physiker Rudolf Clausius verknüpfte 1854 die Ideen von Mayer und Carnot. Er zeigte, dass beim Betreiben einer Dampfmaschine immer Wärme von einem wärmeren Reservoir in ein kälteres Reservoir fließt und damit die Grundthese von Carnot korrekt ist. Jedoch bleibt die Wärmeenergie nicht – wie Carnot annahm – konstant, sondern sie wird zum Teil in mechanische Arbeit umgewandelt. Clausius stellte fest, dass die Wärmeenergie einer Maschine (Dampfmaschine) immer nur zu einem Teil in mechanische Arbeit umgewandelt werden kann; der andere Teil der Energie wird an die Umgebung abgegeben. Der Wirkungsgrad einer Maschine gibt das Umwandlungsverhältnis von gewonnener mechanischer Energie zur zugeführten Wärme an. Clausius Erkenntnis bildet den Zweiten Hauptsatz der Thermodynamik: „Es gibt keine periodisch arbeitende funktionierende Maschine, die nichts anderes tut, als Wärme in mechanische Arbeit zu verwandeln.“[5] Die Wärmemenge, die nicht zur mechanischen Arbeit genutzt werden kann, wird an die Umgebung abgegeben. Diese nicht nutzbare Wärmemenge verknüpfte Clausius mit der entsprechenden Temperatur zu einer neuen Funktion, der Entropie. Alle natürlichen Energieumwandlungsprozesse enthalten einen irreversiblen Entropieanteil, bei dem nicht genutzte Wärme an die Umgebung abgegeben wird. Entropie bedeutet eine „nach innen gekehrte, d. h. nicht mehr verwandlungsfähige oder nutzbare Energie.“[6] Später faßte Boltzmann, recht anschaulich, die Entropie als Maß der Unordnung der Bewegungen eines Systems auf.[7] Nur in einem abgeschlossenen System und bei einer reversiblen Zustandsänderung bleibt die Entropiedifferenz zwischen Anfangs- und Endzustand gleich Null.
Der französische Chemiker Marcelin Berthelot nahm als Triebkraft für eine chemische Reaktion die sich dabei entwickelnde Wärme an (1862).
Hermann Helmholtz verknüpfte die elektrische Energie bei Batterien mit der chemischen Energie und der Wärmeenergie. Er entwickelte in seiner Abhandlung „Ueber die Erhaltung der Kraft“ unabhängig von Mayer den Energieerhaltungssatz.
Helmholtz befasste sich in späteren Jahren mit energetischen Fragen bei chemischen Reaktionen. Helmholtz gab Berthelot recht, dass bei vielen chemischen Umwandlungen Wärme frei wird; es gab jedoch auch Umwandlungen, bei denen Kälte erzeugt wurde. Helmholtz unterteilte in seiner Abhandlung Die Thermodynamik chemischer Vorgänge[8] die Energie bei Stoffumwandlungen in freie und gebundene Energie.[9] Die innere Energie und die Freie Energie verknüpfte Helmholtz mit dem Produkt aus Entropie und Temperatur. Stoffumwandlungen sind nach Helmholtz nur möglich, wenn die Freie Energie abnimmt. Auch der amerikanische Physikochemiker Josiah Willard Gibbs kam nahezu zeitgleich zwischen 1875 und 1878 zu ähnlichen Überlegungen wie Helmholtz. Die Beziehung zwischen Enthalpiedifferenz abzüglich dem Produkt aus Entropiedifferenz und Temperatur bezeichnet man als Differenz der Freien Enthalpie. Die Beziehung heißt zu Ehren der beiden Wissenschaftler Gibbs-Helmholtz-Gleichung. Mit dieser Gleichung kann der Chemiker Aussagen über eine stoffliche Umsetzung von Molekülen machen. Er kann die nötigen Temperaturen und Konzentrationen von chemischen Umsetzungen berechnen.
Neben der klassischen Thermodynamik wurde die kinetische Gastheorie entwickelt. Gase bestehen danach aus Teilchen, Atomen oder Molekülen, die sich zwischen relativ seltenen Stößen frei im leeren Raum bewegen. Bei Temperaturerhöhung bewegen sich die Teilchen schneller und üben durch häufigere und heftigere Stöße einen stärkeren Druck auf die Gefäßwände aus. Wichtige Vertreter dieser Theorie waren August Krönig (1822–1879), Rudolf Clausius, James Clerk Maxwell und Ludwig Boltzmann.[10] Maxwell und Boltzmann nutzten die Wahrscheinlichkeitsrechnung, um thermodynamische Größen auf molekularer Basis zu beschreiben.
Im Jahre 1999 wurde von den Physikern Elliott Lieb und Jakob Yngvason eine axiomatische Systematik vorgestellt, bei der die Definition der Entropie auf dem Konzept der adiabatischen Erreichbarkeit beruht und auf einer streng mathematischen Basis in Form von 15 Axiomen steht. Dabei ist die Temperatur nur noch eine aus der Entropie als Grundgröße abgeleitete Größe. Das Konzept der adiabatischen Erreichbarkeit basiert auf einer axiomatischen Begründung von Constantin Carathéodory aus dem Jahr 1909. Da diese Theorie auf die Ergebnisse keine Auswirkungen hat, hat sie in die Praxis bisher keinen – und in die Lehre nur ausnahmsweise – Eingang gefunden.
Aufgrund der relativ langen Historie der Thermodynamik und der breiten Anwendungsgebiete verwenden die Beschreibungen in der technischen Thermodynamik (z. B. bei der Beschreibung eines Verbrennungsmotors oder eines Kühlschranks), der chemischen Thermodynamik (z. B. bei der Beschreibung einer chemischen Reaktion) und der statistischen Thermodynamik (z. B. bei der Beschreibung von geordneten Quantenzuständen in Festkörpern) oft deutlich unterschiedliche Formalismen.
Kurze Zusammenfassung der Hauptsätze
0. Hauptsatz: Stehen zwei Systeme jeweils mit einem dritten im thermodynamischen Gleichgewicht, so stehen sie auch untereinander im Gleichgewicht.
1. Hauptsatz: Die Energie eines abgeschlossenen Systems ist konstant.
2. Hauptsatz: Thermische Energie ist nicht in beliebigem Maße in andere Energiearten umwandelbar.
3. Hauptsatz: Der absolute Nullpunkt der Temperatur ist unerreichbar.
„Nullter“ Hauptsatz
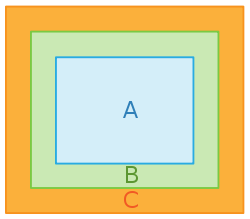 Drei thermodynamische Systeme A, B und C; Näheres
Drei thermodynamische Systeme A, B und C; NäheresWenn ein System A sich mit einem System B sowie B sich mit einem System C im thermischen Gleichgewicht befindet, so befindet sich auch A mit C im thermischen Gleichgewicht.
Anders formuliert: das Gleichgewicht ist transitiv. Dies erlaubt es, eine neue Zustandsgröße, die empirische Temperatur θ, einzuführen, so dass zwei Systeme genau dann die gleiche Temperatur haben, wenn sie sich im thermischen Gleichgewicht befinden. Beispiel: Ein Thermometer ist selbst ein System und soll als B bezeichnet werden. Wenn B die gleiche Temperatur für ein System A, wie auch für ein System C anzeigt, lässt sich daraus schließen, dass auch A und C untereinander im thermischen Gleichgewicht stehen werden, wenn man sie in Kontakt bringt. Dieses Gesetz wurde erst nach den drei anderen Hauptsätzen formuliert. Da es aber eine wichtige Basis bildet, wurde es später als „nullter“ Hauptsatz bezeichnet.
Allerdings ist im Gravitationsfeld zu beachten, dass das Gleichgewicht bei im Allgemeinen verschiedenen Temperaturen zwischen den Systemen A, B und C liegt, denn die Photonen der Schwarzkörperstrahlung erfahren im Gravitationsfeld aufgrund des Äquivalenzprinzips eine Rot-/Blau-Verschiebung; durch die Zeitdilatation werden sie in unterschiedlichen Höhen mit verschiedenen Raten emittiert. Zudem sind deren Flugbahnen gekrümmt, so dass nicht alle von unten startenden Photonen auch oben ankommen können. All diese Effekte bewirken eine mit der Höhe abnehmende Temperatur. Auf der Erde beträgt dieser Effekt aber nur 1,6·10-14 K/m und ist daher unmessbar klein. Bei einem Neutronenstern ist er aber nicht vernachlässigbar.
Erster Hauptsatz
Bilanz für das geschlossene thermodynamische System
Der erste Hauptsatz der Thermodynamik ist aus dem Satz der Energieerhaltung abgeleitet: jedes System besitzt eine innere Energie U (= extensive Zustandsgröße). Diese kann sich nur durch den Transport von Energie in Form von Arbeit W und/oder Wärme Q über die Grenze des Systems ändern, das heißt:
Dabei ist W die Summe aus der Volumenarbeit und der im System dissipierten Arbeit (z. B. Reibungsarbeit). Die Gleichung gilt für das ruhende System. Beim bewegten System kommen die äußeren Energien Ea (potentielle und kinetische Energie) hinzu:
Die Energie eines abgeschlossenen Systems bleibt unverändert. Verschiedene Energieformen können sich demnach ineinander umwandeln, aber Energie kann weder aus dem Nichts erzeugt noch kann sie vernichtet werden. Deshalb ist ein Perpetuum Mobile erster Art unmöglich (kein System verrichtet Arbeit ohne Zufuhr einer anderen Energieform und/oder ohne Verringerung seiner inneren Energie).
Eine Einschränkung der Umwandelbarkeit von Wärme in Arbeit ergibt sich erst aus dem zweiten Hauptsatz der Thermodynamik.
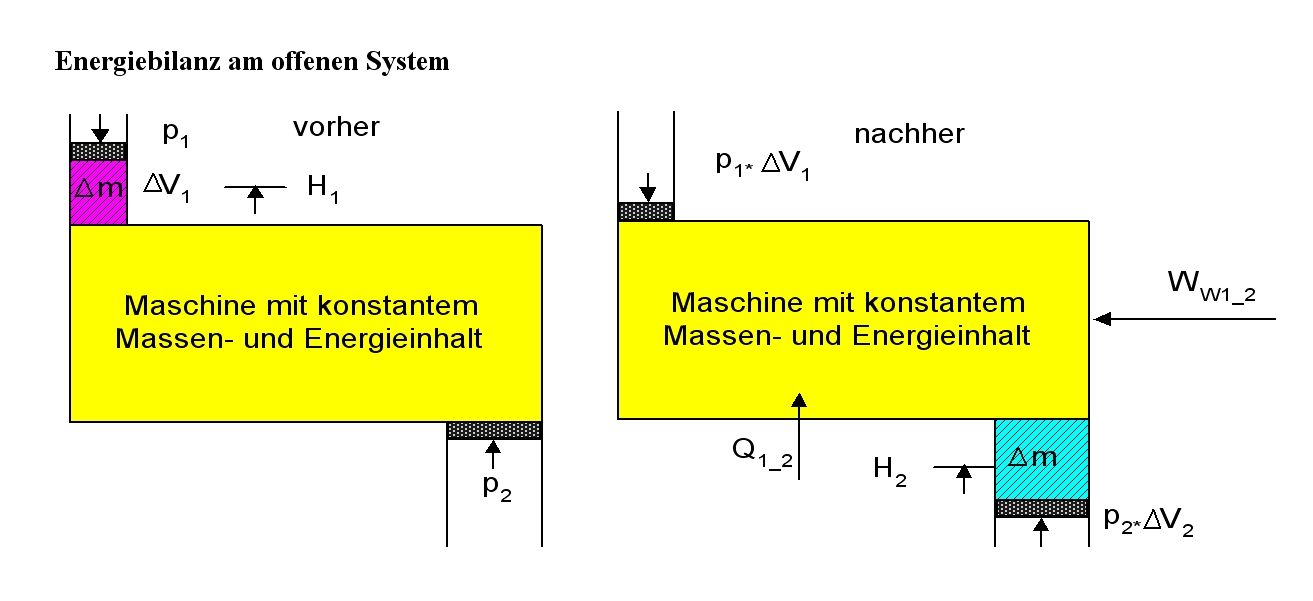
Energiebilanz für ein beliebiges offenes System
Auf das offene System angewendet, wird der erste Hauptsatz mathematisch anders formuliert. Beim offenen System fließen über die bestimmte Systemgrenze zusätzlich zur mechanischen Arbeit an der verschiebbaren Systemgrenze (Volumenänderungsarbeit z. B. am Kolben in einem Zylinder) die Verschiebearbeiten der Massenströme am Ein- und Austritt. Sie sind das Produkt aus Druck und Volumen. Statt mit der inneren Energie wird beim offenen System deshalb mit den Enthalpien bilanziert, die diesen Term enthalten. Es ist:
 bzw.
bzw. 
Die Bilanz für ein instationäres System, bei dem sowohl Masseinhalt als auch Energieinhalt sich zeitlich ändern, lautet:
Dabei sind:
 : die zeitliche Änderung des Energieinhalts im System (Energieinhalt = innere Energie + kinetische Energie + potentielle Energie).
: die zeitliche Änderung des Energieinhalts im System (Energieinhalt = innere Energie + kinetische Energie + potentielle Energie). : der Wärmestrom über die Systemgrenze.
: der Wärmestrom über die Systemgrenze. : der Arbeitsstrom (technische Arbeit) über die Systemgrenze.
: der Arbeitsstrom (technische Arbeit) über die Systemgrenze. : der Massenstrom in das System.
: der Massenstrom in das System. : der Massenstrom aus dem System
: der Massenstrom aus dem System- h: die spezifische Enthalpie
 : die spezifische potentielle Energie (mit z = Höhe über dem Bezugsniveau und g = Schwerebeschleunigung)
: die spezifische potentielle Energie (mit z = Höhe über dem Bezugsniveau und g = Schwerebeschleunigung) : die spezifische kinetische Energie (mit c = Geschwindigkeit).
: die spezifische kinetische Energie (mit c = Geschwindigkeit).
Sonderfälle und Vereinfachungen:
- Geschlossenes System:
 (siehe oben)
(siehe oben)
- stationär:
 und
und 
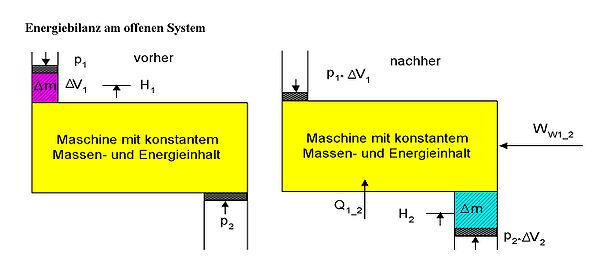 Energiebilanz am offenen stationären System. Es wird ein kleiner Zeitraum Δt betrachtet, in dem die Masse Δm mit dem Zustand 1 in das System fließt und dieses im Zustand 2 wieder verlässt. Der Massenstrom ist dann Δm / Δt. Die Verschiebarbeiten am Eintritt und Austritt werden jeweils mit der inneren Energie in der Enthalpie zusammengefasst.
Energiebilanz am offenen stationären System. Es wird ein kleiner Zeitraum Δt betrachtet, in dem die Masse Δm mit dem Zustand 1 in das System fließt und dieses im Zustand 2 wieder verlässt. Der Massenstrom ist dann Δm / Δt. Die Verschiebarbeiten am Eintritt und Austritt werden jeweils mit der inneren Energie in der Enthalpie zusammengefasst. oder:
oder:
- zusätzlich adiabat (z. B. Dampfturbine):

Dabei ist P die Wellenleistung der Maschine. Da vom System abgegebene Energien in der Thermodynamik negativ definiert sind, wird die Leistung einer Turbine aus dieser Gleichung negativ. In der Praxis wird das Vorzeichen deshalb gewechselt. In vereinfachten Berechnungen vernachlässigt man auch die äußeren Energien. Dann lässt sich bei bekannten Zuständen am Eintritt und Austritt die spezifische Leistung direkt als Ordinatendifferenz aus dem h-s-Diagramm ablesen.
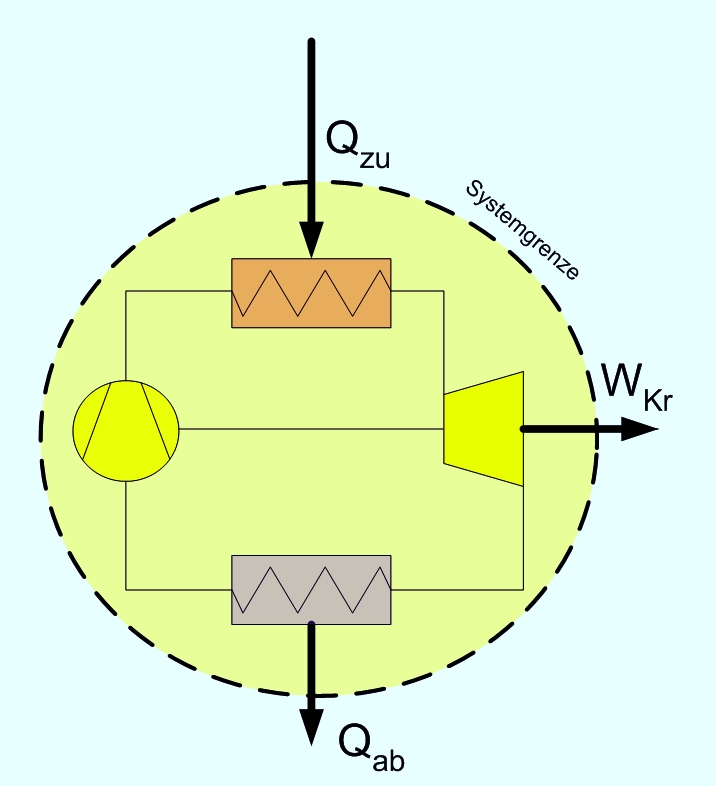
Energiebilanz für Kreisprozesse
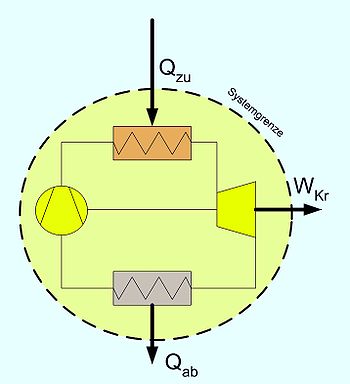 1. Hauptsatz für den Kreisprozess. Ein Kreisprozess kann als geschlossenes, inhomogenes System betrachtet werden, über dessen Grenze nur Wärme und Arbeit fließt. Als Beispiel ist hier ein Gasturbinenprozess mit Wärmeübertragern gezeichnet.
1. Hauptsatz für den Kreisprozess. Ein Kreisprozess kann als geschlossenes, inhomogenes System betrachtet werden, über dessen Grenze nur Wärme und Arbeit fließt. Als Beispiel ist hier ein Gasturbinenprozess mit Wärmeübertragern gezeichnet.Da nach dem Durchlaufen eines Kreisprozesses das Arbeitsmedium zum Ausgangszustand zurückkehrt, vereinfacht sich die Bilanz, es entfallen die Änderungen der Zustandsgrößen, und es verbleiben die Prozessgrößen Wärme und Arbeit. Wie noch im Zusammenhang mit dem 2. Hauptsatz erläutert wird, kann nicht nur Wärme zugeführt werden, die komplett in Arbeit umgewandelt wird, sondern es muss auch Wärme abgeführt werden. Die einfache Bilanzgleichung lautet:
Dabei summiert das Kreisintegral alle Wärmeströme auf. Sie sind positiv, wenn sie in das System eintreten und negativ, wenn sie es verlassen. WKr ist die gesamte Arbeit des Zyklus. Sie ist negativ, wenn sie abgegeben wird.
Die Beziehung wird auch oft mit den Wärmebeträgen geschrieben:
 ,
,
wobei die Wärmeabfuhr deutlicher erkennbar wird.
Schließlich sollte auch der thermische Wirkungsgrad einer Kraftmaschine
noch genannt werden, der den Nutzen (die Kreisprozessarbeit) ins Verhältnis zum Aufwand setzt (die zugeführte Wärme, die meist in Form von Brennstoff bezahlt werden muss). Die abgeführte Wärme wird in der Regel von der Umgebung aufgenommen.
Zweiter Hauptsatz
Der Zweite Hauptsatz ermöglicht die Einführung der thermodynamischen Entropie als Zustandsgröße zur numerischen und anschaulichen Beschreibung von Prozessen (vgl. T-s-Diagramm) und auch die Definition der thermodynamischen Temperatur. Er schränkt die Aussage des ersten Hauptsatzes über die Gleichwertigkeit von Wärme und Arbeit ein und ist damit eines der Fundamente der Thermodynamik, wird aber im Rahmen dieser Theorie nicht begründet. Erst im Rahmen der Statistischen Mechanik wird er mit den übrigen Theorien der Physik verknüpft: Je nach philosophischem Standpunkt bekommt er dort eine stochastische Formulierung oder wenigstens eine wahrscheinlichkeitsbezogene Begründung. Das Wort Wärmestrahlung bei Bemerkungen zum Zweiten Haupzsatz bezieht sich immer auf die Nettostrahlung - zu Clausius Zeiten verstand man unter Wärmestrahlung immer die Nettostrahlung. Das heutige Verständnis von Wärmestrahlung existierte damals noch nicht.
Die verschiedenen Aussagen
Der Zweite Hauptsatz der Thermodynamik in der Formulierung von Clausius lautet:
Es gibt keine Zustandsänderung, deren einziges Ergebnis die Übertragung von Wärme von einem Körper niederer auf einen Körper höherer Temperatur ist.
Einfacher ausgedrückt: Wärme kann nicht von selbst von einem Körper niedriger Temperatur auf einen Körper höherer Temperatur übergehen. Diese Aussage scheint zunächst überflüssig zu sein, denn sie entspricht der alltäglichen Erfahrung, wie die über die Anziehungskraft der Erde. Dennoch ist sie äquivalent zu allen weiteren, weniger „selbstverständlichen“ Aussagen, denn alle Widersprüche zu den anderen Aussagen lassen sich auf einen Widerspruch zu dieser zurückführen.
Der Zweite Hauptsatz der Thermodynamik in der Formulierung von Kelvin und Planck lautet:
„Es ist unmöglich, eine periodisch arbeitende Maschine zu konstruieren, die weiter nichts bewirkt als Hebung einer Last und Abkühlung eines Wärmereservoirs.“[11]
Dem ersten Hauptsatz würde die Annahme nicht widersprechen, dass es möglich sei, einer – wie immer auch gearteten – Kraftmaschine einen stetigen Wärmestrom zuzuführen, den diese vollständig als mechanische oder elektrische Leistung abgibt. Eine solche Maschine wird als Perpetuum mobile zweiter Art bezeichnet. Eine entsprechende Formulierung des zweiten Hauptsatzes lautet:
Ein Perpetuum mobile zweiter Art ist unmöglich.
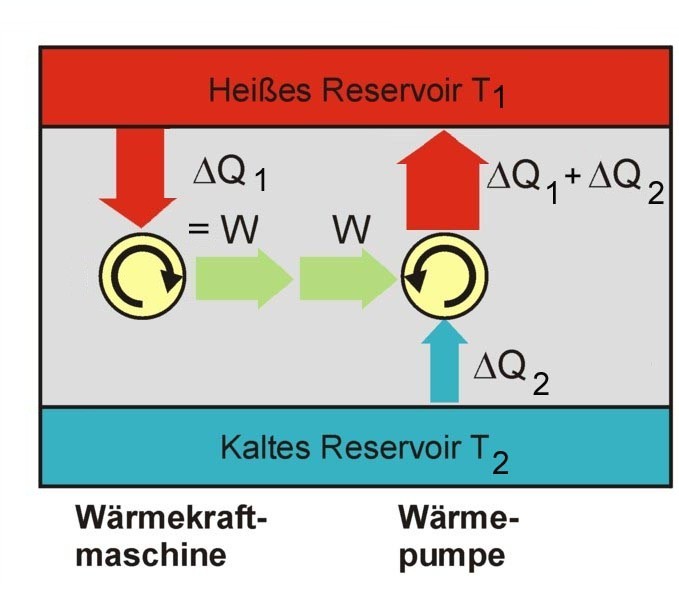
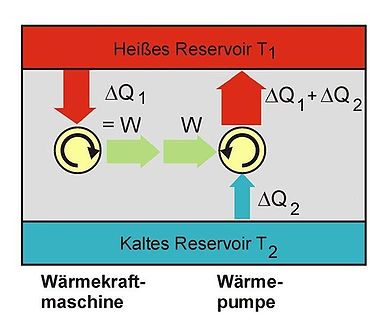 Die Kraftmaschine links im Bild (Kreisprozess im Uhrzeigersinn) wäre, wenn es sie gäbe, ein Perpetuum mobile 2. Art. Die gesamte Anordnung mit der von dieser Maschine angetriebenen Wärmepumpe würde bewirken, dass innerhalb dieses Systems ohne Einwirkung von außen Wärme vom kalten zum warmen Reservoir fließt.
Die Kraftmaschine links im Bild (Kreisprozess im Uhrzeigersinn) wäre, wenn es sie gäbe, ein Perpetuum mobile 2. Art. Die gesamte Anordnung mit der von dieser Maschine angetriebenen Wärmepumpe würde bewirken, dass innerhalb dieses Systems ohne Einwirkung von außen Wärme vom kalten zum warmen Reservoir fließt.Nimmt man an, es gäbe diese von einer Wärmesenke zur Wärmeabfuhr unabhängige Kraftmaschine, so könnte damit der Umgebung, z. B. dem Meerwasser, Wärme entzogen und in mechanische Arbeit umgewandelt werden. Man könnte damit auch gemäß dem Bild rechts die Wärme aus einem Reservoir oder Behälter entziehen und mit der umgewandelten Energie eine Wärmepumpe antreiben, die mit einem reversiblen Carnot-Prozess Wärme aus einem anderen Behälter mit niedrigerer Temperatur in den ersteren mit höherer Temperatur fördert. Die in den wärmeren Behälter eingespeiste Wärmemenge wäre dann größer als die von der Kraftmaschine aufgenommene, weil die abgegebene Energie der Wärmepumpe aus der Summe von aufgenommener Wärme und Antriebsarbeit besteht. Denkt man sich die Systemgrenze um beide Maschinen einschließlich der beiden Wärmebehälter gezogen, so wäre innerhalb dieses abgeschlossenen Systems – also ohne Energie- und Stoffaustausch mit der Umgebung – letztlich Wärme von einem kälteren zu einem wärmeren Körper geflossen. Dies ist ein Widerspruch zur ersten Aussage. Prinzipiell derselbe Widerspruch ergibt sich aber auch mit der Annahme, man könnte eine Kraftmaschine bauen, die einen größeren Wirkungsgrad aufweist als eine mit einem Carnot-Prozess arbeitende Maschine. Auch diese Maschine würde dem wärmeren Behälter weniger Wärme entnehmen als die von ihr angetriebene Carnot-Wärmepumpe dort einspeist. Die entsprechende Aussageform des zweiten Hauptsatzes lautet:
Es gibt keine Wärmekraftmaschine, die bei gegebenen mittleren Temperaturen der Wärmezufuhr und Wärmeabfuhr einen höheren Wirkungsgrad hat als der aus diesen Temperaturen gebildete Carnot-Wirkungsgrad.
Die Nennung der mittleren Temperaturen ist deshalb von Bedeutung, weil in der Regel durch Wärmezufuhr oder Wärmeentnahme ein Wärmereservoir seine Temperatur ändert.
Unmittelbar in diesem Zusammenhang lässt sich weiter formulieren:
Alle reversiblen Wärme-Kraft-Prozesse mit gleichen mittleren Temperaturen der Wärmezufuhr und Wärmeabfuhr haben denselben Wirkungsgrad wie der entsprechende Carnot-Prozess.
und:
Alle irreversiblen Wärme-Kraft-Prozesse haben einen geringeren Wirkungsgrad.
Mit den in der modernen Thermodynamik festgelegten Begriffsdefinitionen (Wärme, Arbeit, Innere Energie, Zustandsgröße, Prozessgröße, adiabat…) und mit der systematischen Einteilung der Systeme kann über die von Clausius eingeführte Zustandsgröße Entropie eine für alle geschlossenen Systeme und Prozesse in offenen Systemen allgemein gültige Aussage des zweiten Hauptsatzes in mathematischer Form gegeben werden (Bei offenen Systemen bezieht sich die Bilanz auf ein Fluidteilchen, das sich durch das System hindurch bewegt und als geschlossenes bewegtes System betrachtet werden kann (siehe oben) ).
Dabei ist δWdiss die innerhalb des Systems dissipierte Arbeit (Arbeit, die nicht nach außen gelangt, sondern infolge von Reibungs-, Drosselungs- oder Stoßvorgängen die innere Energie erhöht). Sie ist immer positiv. Man bezeichnet den entsprechenden Term in der Gleichung als „produzierte Entropie“ – im Gegensatz zum ersten Term, der „transportierte Entropie“ genannt wird und auch negativ sein kann.
Für das adiabate System mit δQ = 0 ergibt sich daraus:
In einem geschlossenen adiabaten System kann die Entropie nicht abnehmen, sie nimmt in der Regel zu. Nur bei reversiblen Prozessen bleibt sie konstant.
Auch hier ist die Äquivalenz mit der ersten Aussage von Clausius leicht zu erkennen. Ein selbsttätiger Wärmefluss vom kälteren zum wärmeren Behälter in der oben skizzierten Anordnung würde bedeuten, dass die Entropie des kälteren Behälters (geringere Temperatur T im Nenner) stärker abnimmt, als die des wärmeren zunimmt, d. h. die gesamte Entropie im System abnimmt, was nicht möglich ist.
Alle spontan ablaufenden Prozesse sind irreversibel. Dort findet immer eine Entropiezunahme statt. Beispiele sind die Vermischung von zwei unterschiedlichen Gasen und der Wärmefluss von einem heißen zu einem kalten Körper ohne Gewinnung von Arbeit. Die Wiederherstellung des (oft „geordneter“ genannten) Anfangszustandes erfordert dann den Einsatz von Energie oder Information (siehe maxwellscher Dämon). Reversible Prozesse sind nicht mit einer Erhöhung der Gesamtentropie verbunden und laufen daher auch nicht spontan ab. Durch die theoretische Beschreibung spontan ablaufender Prozesse zeichnet der Zweite Hauptsatz der Thermodynamik eine Richtung der Zeit aus, die mit unserer intuitiven Erfahrungswelt übereinstimmt (vgl. das Beispiel weiter unten).
Mit den beschriebenen Zusammenhängen ist auch der folgende Satz eine Aussageform des zweiten Hauptsatzes:
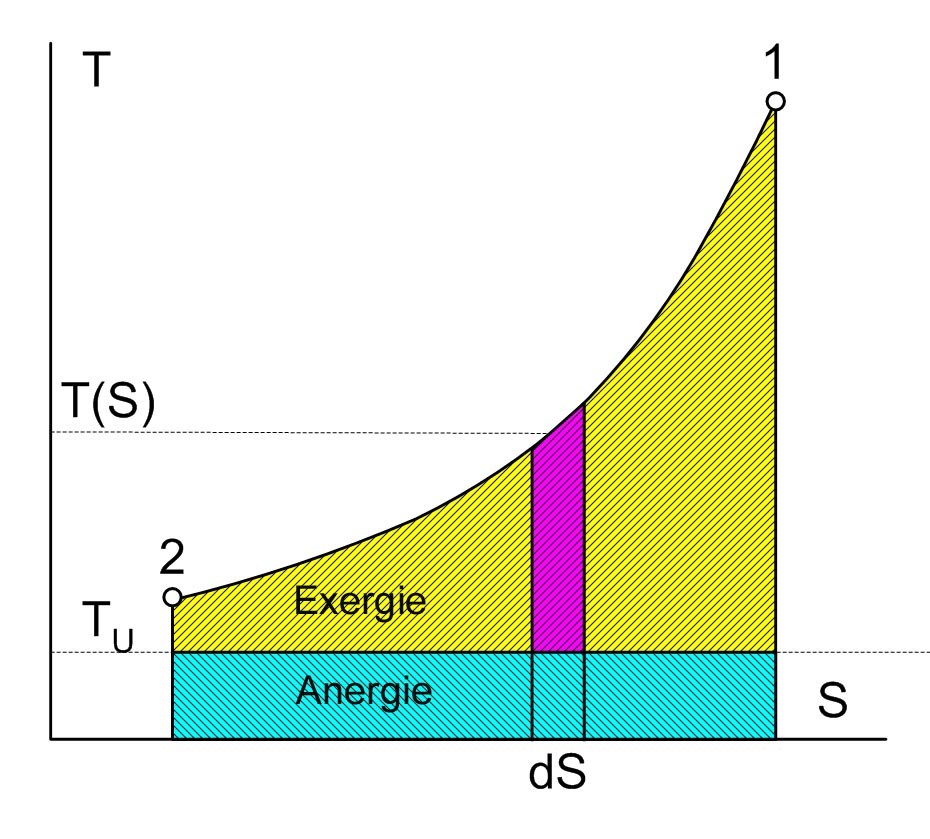
Die Thermische Energie eines Systems besteht aus einem Anteil Exergie und einem Anteil Anergie, wobei der exergetische Anteil verschwindet, wenn das System in den Umgebungszustand übergeführt wird
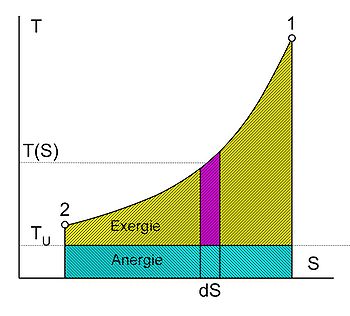 Exergie und Anergie der Wärme (Thermische Energie = Anergie + Exergie)
Exergie und Anergie der Wärme (Thermische Energie = Anergie + Exergie)Die Exergie ist der umwandelbare Anteil thermischer Energie in andere Energieformen. Wird ein Körper bzw. System mit einem Zustand, der von dem der Umgebung abweicht, reversibel in den Umgebungszustand gebracht, so wird seine Exergie als Arbeit abgegeben. Die Wärme, die ein Körper (z. B. ein heißes Rauchgas im Kessel eines Kraftwerks) abgibt, wenn es sich auf Umgebungstemperatur abkühlt, kann theoretisch über eine Folge von differenziellen Carnot-Prozessen, wie im Bild rechts dargestellt, zur Umwandlung in Arbeit genutzt werden. Der exergetische Anteil ergibt sich durch Aufsummieren der differenziellen (pinkfarbenen) Flächenanteile oberhalb der Umgebungstemperatur TU.
Die Wärmesenke für diese Prozesse zur Aufnahme der Anergie (blauer Flächenanteil unterhalb TU) ist die Umgebung. Herrscht bei einem Gas im Ausgangszustand gegenüber dem Umgebungszustand nicht nur eine höhere Temperatur, sondern auch ein höherer Druck, so besteht die gesamte Exergie nicht nur aus dem exergetischen Anteil der Wärme, sondern zusätzlich aus einem Anteil Volumenarbeit.
Der thermische Wirkungsgrad der realen Wärmekraftmaschine ist also immer kleiner als 1 und – bedingt durch die von den Maschinen vorgegebene Prozessführung und die unvermeidlichen dissipativen Effekte – auch immer kleiner als der der idealen Wärmekraftmaschine:
wobei TU die Umgebungstemperatur ist und
 die mittlere Temperatur der Wärmezufuhr. Sie ergibt sich, wenn die gelbe Fläche der Exergie durch ein flächengleiches Rechteck oberhalb der Linie der Umgebungstemperatur ersetzt wird.
die mittlere Temperatur der Wärmezufuhr. Sie ergibt sich, wenn die gelbe Fläche der Exergie durch ein flächengleiches Rechteck oberhalb der Linie der Umgebungstemperatur ersetzt wird.Der Zweite Hauptsatz hat somit erhebliche technische Auswirkungen. Da viele Maschinen, die mechanische Energie liefern, diese über einen Umweg aus thermischer Energie erzeugen (z. B. Dieselmotor: Chemische Energie
 thermische Energie mechanische Energie), gelten für ihre Wirkungsgrade immer die Beschränkungen des 2. Hauptsatzes. Im Vergleich dazu bieten Wasserkraftanlagen, die bei der Umwandlung keine Zwischenstufe über thermische Energie benötigen, erheblich höhere Wirkungsgrade.
thermische Energie mechanische Energie), gelten für ihre Wirkungsgrade immer die Beschränkungen des 2. Hauptsatzes. Im Vergleich dazu bieten Wasserkraftanlagen, die bei der Umwandlung keine Zwischenstufe über thermische Energie benötigen, erheblich höhere Wirkungsgrade.Beispiele
1. Beispiel:
Ein kräftefreies Gas verteilt sich immer so, dass es das zur Verfügung stehende Volumen vollständig und gleichmäßig ausfüllt. Warum das so ist, versteht man, wenn man den gegenteiligen Fall betrachtet. Man stelle sich eine luftdichte Kiste in der Schwerelosigkeit vor, in der sich ein einziges Partikel bewegt. Die Wahrscheinlichkeit, dieses bei einer Messung in der linken Hälfte der Kiste zu finden, ist dann genau 1/2. Befinden sich dagegen zwei Partikel in der Kiste, dann ist die Wahrscheinlichkeit, beide in der linken Hälfte anzutreffen, nur noch 1/2 · 1/2 = 1/4, und bei N Partikeln entsprechend 0,5N. Die Anzahl der Atome in einem Gas ist unvorstellbar hoch. In einem Volumen von einem Kubikmeter bei normalem Druck liegt sie in der Größenordnung von rund 3·1025 Teilchen. Die daraus resultierende Wahrscheinlichkeit, dass sich das Gas in der Kiste spontan in einer Hälfte konzentriert, ist so gering, dass ein solches Ereignis vermutlich niemals eintreten wird.
Wie aus den zeitlich umkehrbaren mikroskopischen Gleichungen der klassischen Mechanik (ohne Reibung) die symmetriebrechende makroskopische Gleichung folgt, wird in der statistischen Mechanik geklärt. Zudem erhält die Entropie dort eine anschauliche Bedeutung: sie ist ein Maß der Unordnung eines Systems bzw. der im System enthaltenen Informationen. Allerdings verliert der Zweite Hauptsatz in der statistischen Mechanik seinen Status als „streng gültiges“ Gesetz, sondern wird dort als Gesetz betrachtet, bei dem Ausnahmen auf makroskopischen Ebenen zwar prinzipiell möglich, aber gleichzeitig so unwahrscheinlich sind, dass sie praktisch nicht vorkommen. Auf mikroskopischer Ebene betrachtet führen z. B. kleine statistische Fluktuationen um den Gleichgewichtszustand auch bei abgeschlossenen Systemen dazu, dass die Entropie ebenfalls etwas um den Maximalwert fluktuiert und dabei auch abnehmen kann.
2. Beispiel:
Folgendes Beispiel soll die Bedeutung des Begriffs „Zustand“ in der Thermodynamik hervorheben und den Unterschied von Zustandsgrößen und Nicht-Zustandsgrößen illustrieren.
Wir betrachten dazu einen mittels eines beweglichen Kolbens abgeschlossenen Zylinder, der mit N0 Molen eines idealen Gases gefüllt ist. Der Zylinder befindet sich in Wärmekontakt mit einem Wärmebad der Temperatur T0.
Zunächst befindet sich das System im Zustand 1, charakterisiert durch (T0,V1,N0); dabei ist V1 das Volumen des Gases. Ein Prozess soll das System in den Zustand 2 gegeben durch (T0,V2,N0) mit V2 > V1 bringen. Temperatur und Stoffmenge bleiben also konstant und das Volumen vergrößert sich.
Wir diskutieren zwei verschiedene isotherme Prozesse, die das leisten: (1) eine instantane Expansion (Joule-Thomson-Expansion) und (2) eine quasistatische Expansion.
Bei Prozess (1) wird der Kolben „unendlich“ schnell herausgezogen (man kann den Prozess auch folgendermaßen realisieren: ein Gefäß mit einem Volumen V2 ist durch eine herausnehmbare Wand in zwei Teilbereiche geteilt, wobei einer das Volumen V1 besitzt und mit dem idealen Gas gefüllt ist. Der andere Teilbereich ist evakuiert. Der Prozess ist dann durch das Herausziehen der Zwischenwand gegeben). Dabei leistet das Gas keine Arbeit, es ist also δW = 0. Experimentell zeigt sich, dass sich die Energie des Gases nicht ändert (der mittlere Geschwindigkeitsbetrag der Gasteilchen bleibt gleich), daher ist auch die Wärme („in Form von Wärme zugeführte Energie“) gleich Null: δQ = 0. Zusammengefasst: Bei Prozess (1) ist die Energie von Anfangs- und Endzustand gleich. Die Energieformen Arbeit und Wärme verschwinden.
Bei Prozess (2) wird der Kolben sehr langsam herausgezogen und dadurch das Volumen vergrößert. Das Gas leistet Arbeit, es ist δW < 0. Da die Energie von Anfangs- und Endzustand aber dieselbe ist (die Energie ist eine Zustandsgröße und hängt nicht von der Prozessführung ab!), muss nach dem ersten Hauptsatz bei dem Prozess Energie in Form von Wärme zugeführt werden: δQ = − δW > 0. Zusammengefasst: Bei Prozess (2) ist die Energie von Anfangs- und Endzustand (ebenfalls) gleich. Das System leistet Arbeit („verliert Energie in Form von Arbeit“) und erhält vom Wärmebad Energie in Form von Wärme.
Insgesamt sieht man also, dass die Energieformen Wärme und Arbeit von der konkreten Realisierung des Prozesses abhängen. In der Thermodynamik benutzt man die Bezeichnung d für Differentiale von Zustandsgrößen und δ für infinitesimal kleine Änderungen von Nicht-Zustandsgrößen. Ein System besitzt in einem Zustand eine bestimmte Energie, Entropie, Volumen etc., aber keine Wärme oder Arbeit!
Noch eine Anmerkung: Bei Prozess (1) verlässt das System den thermodynamischen Zustandsraum. Die Zustände, die das System zwischen Anfangs- und Endzustand einnimmt, sind keine thermodynamischen Gleichgewichtszustände. Daher sind die Differentiale im 1. Hauptsatz nicht definiert. Dieser gilt jedoch auch für endliche Differenzen. Die obige Betrachtung ist auch für einen nicht-quasistatischen Prozess korrekt.
Zusammenfassung der Aussagen des zweiten Hauptsatzes
- Wärme kann nicht von selbst von einem Körper niedriger Temperatur auf einen Körper höherer Temperatur übergehen.
- Wärme kann nicht vollständig in Arbeit umgewandelt werden. Dies wäre eine Realisierung eines Perpetuum Mobile zweiter Art.
- Der Wirkungsgrad des Carnot-Prozesses kann nicht übertroffen werden.
- Alle spontan (in eine Richtung) ablaufenden Prozesse sind irreversibel.
- Alle Prozesse, bei denen Reibung stattfindet, sind irreversibel.
- Ausgleichs- und Mischungsvorgänge sind irreversibel.
- In einem geschlossenen adiabaten System kann die Entropie nicht geringer werden.
- Das Gleichgewicht isolierter thermodynamischer Systeme ist durch ein Maximalprinzip der Entropie ausgezeichnet.
Gültigkeit
Der Zweite Hauptsatz der Thermodynamik stellt eine Erfahrungstatsache dar. Es ist bis heute nicht gelungen, dieses fundamentale Gesetz der klassischen Physik in seiner allgemeinen Gültigkeit für beliebige makroskopische Systeme ausgehend von der Grundgleichung der Quantentheorie, der Vielteilchen-Schrödingergleichung, zu beweisen. Dies gilt selbstverständlich auch umgekehrt: Die Schrödingergleichung stellt eine Erfahrungstatsache dar. Es ist bis heute nicht gelungen, die allgemeine Gültigkeit dieses fundamentalen Gesetzes quantenmechanischer Systeme für beliebige makroskopische Systeme, ausgehend von den beiden Hauptsätzen der Physik (und nicht nur der Thermodynamik!), zu beweisen.
Dritter Hauptsatz
Dieser Hauptsatz wurde von Walther Nernst im Jahr 1906 vorgeschlagen und ist auch als Nernst-Theorem bekannt. Er ist quantentheoretischer Natur und äquivalent zur Aussage von der Unerreichbarkeit des Nullpunktes der absoluten Temperatur:
- Es ist nicht möglich, ein System bis zum absoluten Nullpunkt abzukühlen.
Bei der Annäherung der Temperatur an den absoluten Nullpunkt (T = 0K) wird die Entropie S unabhängig von thermodynamischen Parametern. Damit geht S gegen einen festen Grenzwert S0:
Die konstante Entropie bei T = 0K lässt sich als
 darstellen, wobei k die Boltzmann-Konstante ist und Ω0 die Anzahl der möglichen Mikrozustände im Grundzustand (Entartung). Zum Beispiel würde sich für einen n-atomigen Kristall, dessen Atome im Energiegrundzustand zwei mögliche Spineinstellungen haben,
darstellen, wobei k die Boltzmann-Konstante ist und Ω0 die Anzahl der möglichen Mikrozustände im Grundzustand (Entartung). Zum Beispiel würde sich für einen n-atomigen Kristall, dessen Atome im Energiegrundzustand zwei mögliche Spineinstellungen haben,  ergeben.
ergeben.Für alle physikalisch-chemischen Reaktionen, bei denen die teilnehmenden Stoffe am absoluten Nullpunkt als ideale kristalline Festkörper vorliegen, gilt:
Es gibt nur eine Realisierungsmöglichkeit für ideale Festkörper am absoluten Nullpunkt, Ω0 = 1.
Die genannten Aussagen können mit Methoden der Quantenstatistik rigoros bewiesen werden.
Energieberechnungen in der Thermodynamik
Die Energiebilanz hat in der Thermodynamik einen hohen Stellenwert.
Bei einer Phasenumwandlung (fest-flüssig-gasförmig) oder Mischungen (Salz in Wasser, Mischung verschiedener Lösungsmittel) werden Umwandlungsenergien (Schmelzenthalpie, Verdampfungsenthalpie, Sublimationsenthalpie) oder Umwandlungsenthalpien benötigt bzw. werden in umgekehrter Richtung frei. Bei einer chemischen Stoffumwandlung können Reaktionswärmen oder Reaktionsenthalpien frei werden oder müssen umgekehrt zugeführt werden.
Zur Berechnung von frei werdenden Reaktionswärmen bei Stoffumsetzungen wird zunächst die entsprechende Reaktionsgleichung mit den dazugehörigen stöchiometrischen Faktoren aufgestellt. Die Standardbildungsenthalpien der Einzelstoffe sind für 25 °C in Tabellenwerken verzeichnet. Man addiert die Summe der Enthalpien der Produkte entsprechend den stöchiometrischen Faktoren und zieht davon die Enthalpien der Ausgangsstoffe ab (Hess'scher Wärmesatz).
Die Reaktions- oder Umwandlungsenthalpie, die bei einer chemischen Umsetzung oder Phasenumwandlung an die Umgebung abgegeben wird, hat ein negatives Vorzeichen. Ist eine Energiezufuhr von der Umgebung für eine Phasenumwandlung oder eine chemische Umsetzung nötig, so hat diese ein positives Vorzeichen.
Die Zustandsgröße für Enthalpie ist

Die Freie Enthalpie ist

Durch Bildung des totalen Differentials der Freien Enthalpie und der anschließenden Integration lässt sich berechnen, ob eine chemische Umsetzung möglich ist.

Ist die Differenz der Freien Enthalpien (ΔG) der Produkte zu den Ausgangsstoffen (Edukte) negativ, ist eine Phasenumwandlung oder eine Stoffumsetzung möglich. Ist die Differenz der Freien Enthalpie einer Reaktion, einer Phasenumwandlung negativ, erfolgt eine Reaktion – soweit diese nicht kinetisch gehemmt ist – bis zu einem Punkt, an dem ΔG = 0 wird. Das Massenwirkungsgesetz ist ein Spezialfall eines solchen Gleichgewichtes. Ist die Differenz der Freien Enthalpie positiv, so ist eine Reaktion oder Phasenumwandlung unmöglich.
Im Jahr 1869 glaubte Marcellin Berthelot noch, dass nur chemische Umwandlungen möglich seien, bei denen Wärme frei wird.
Mitunter ist eine Umwandlung oder eine Reaktion möglich, obgleich dabei keine Reaktionswärme oder Umwandlungswärme frei wird. Dies liegt am Entropieterm T*ΔS.
Beispiele:
- Bei Lösen von Glaubersalz in Wasser wird die Lösung kälter als die Umgebung. Die Enthalpieterm ist positiv, jedoch nimmt die Unordnung, d. h. die Entropie, durch die Auflösung zu.
- Beim Schmelzen eines Eisblockes wird Wärme zur Phasenumwandlung von fest zu flüssig benötigt.
- Die Wärme des Wassers steigt kaum, obgleich Wärme von der Umgebung zugeführt wird. Die Unordnung, die Entropie der Moleküle ist im flüssigen Zustand größer als im festen Zustand.
- Bei der Umwandlung von Kohle und Kohlendioxid zu Kohlenmonoxid ist die Reaktionsenthalpie positiv. Durch die Reaktionsentropie lässt sich das Gleichgewicht (siehe: Boudouard-Gleichgewicht) bei hoher Temperatur zum Kohlenmonoxid verschieben.
Thermodynamik irreversibler Prozesse
Neben der klassischen Gleichgewichtsthermodynamik wurde im 20. Jahrhundert die Nichtgleichgewichtsthermodynamik oder auch Thermodynamik irreversibler Prozesse entwickelt. Für diese Arbeiten wurden die Nobelpreise der Chemie im Jahr 1968 an Lars Onsager und 1977 an Ilya Prigogine verliehen.
Die klassische Thermodynamik macht über Nichtgleichgewichtsprozesse nur die qualitative Aussage, dass diese nicht umkehrbar sind, beschränkt sich aber in ihren quantitativen Aussagen auf Systeme, die stets global im Gleichgewicht sind bzw. nur inkrementell davon abweichen. Demgegenüber behandelt die Nichtgleichgewichtsthermodynamik Systeme, die sich nicht in einem globalen thermodynamischen Gleichgewicht befinden, sondern davon abweichen. Oft wird jedoch noch ein lokales thermodynamisches Gleichgewicht angenommen.
Ein wichtiges Ergebnis der Nichtgleichgewichtsthermodynamik ist das Prinzip der minimalen Entropieproduktion für offene Systeme, welche nur wenig vom thermodynamischen Gleichgewicht abweichen. Dies ist der Bereich der so genannten linearen irreversiblen Thermodynamik. Sie beschreibt in einem vereinheitlichten formalen Rahmen lineare Zusammenhänge zwischen Flüssen und ihren korrespondierenden Kräften. Diese Kräfte werden normalerweise als Gradienten einer skalaren Größe aufgefasst und die Flüsse durch bekannte lineare Naturgesetze beschrieben, wie zum Beispiel das ohmsche Gesetz (Stromfluss), das Ficksche Gesetz (Diffusion), das Fouriersches Gesetz (Wärmeleitung) oder die Kinetik einer chemischen Reaktion (Reaktionsgeschwindigkeit). Durch die Bilanzierung der Entropie, in die die Produktion der Entropie in dem System und die über die Systemgrenzen fließende Entropie eingehen, lässt sich durch den zweiten Hauptsatz die Invarianz dieser Gesetze zeigen. Für das Beispiel der Wärmeleitung zeigt sich, dass mit der Thermodynamik nur ein Wärmefluss vom heißen zum kalten vereinbar ist, und dass die Wärmeleitfähigkeit immer eine positive Größe sein muss. Durch die mathematische Analyse wird außerdem gezeigt, dass eine thermodynamische Kraft (z. B. Temperaturdifferenz oder Spannungsdifferenz) in einem System einen zusätzlichen indirekten Fluss verursacht (Beispiel: elektrischer Stromfluss verursacht durch Wärmeleitung (Seebeck-Koeffizient), oder Wärmeleitung verursacht einen elektrischen Stromfluss (Peltier-Koeffizient)). Von Lars Onsager wurde gezeigt, dass die Einflüsse zwischen Flüssen und den nicht dazu korrespondierenden Kräften gleich groß ist (Reziprozitätsbeziehungen). Da die Entropiebilanz in einem geschlossen System immer positiv sein muss, folgt zusätzlich: Die Größe der Kreuzeffekte ist immer wesentlich kleiner als die direkten Effekte. Für das Beispiel mit den zwei Kräften gilt, dass die Kreuzeffekte (Peltier Koeffizent und Seebeck Koeffizient) maximal zweimal der Wurzel aus den Produkten der Koeffizienten der beiden direkten Effekte (elektrische und thermische Leitfähigkeit) entspricht.
Weicht ein offenes System stark vom Gleichgewicht ab, kommt die nichtlineare Nichtgleichgewichtsthermodynamik zum Zug. Wichtiges Ergebnis in diesem Bereich ist das Stabilititätskriterium von Ilya Prigogine und Paul Glansdorff, das angibt, unter welchen Bedingungen der Zustand mit der minimalen Entropieproduktion instabil wird und ein System bei gleichzeitigem Entropieexport eine höher geordnete Struktur annehmen kann. In diesem Bereich können also spontan so genannte dissipative Strukturen entstehen, die experimentell bestätigt wurden (beispielsweise Bénard-Zellen). Da in diesem nichtlinearen Bereich auch biologische Prozesse anzusiedeln sind, ist dieses Resultat besonders auch in Hinsicht auf die Entwicklung des Lebens von großer Bedeutung.
Vertreter
- Pierre Prévost (Prévostscher Satz)
- James Prescott Joule
- Nicolas Léonard Sadi Carnot
- Julius Robert von Mayer
- Hermann von Helmholtz
- William Thomson, 1. Baron Kelvin
- James Clerk Maxwell
- Ludwig Boltzmann
- Joseph Louis Gay-Lussac
- Robert Boyle
- Edme Mariotte
- Rudolf Clausius
- Josiah Willard Gibbs
- Guillaume Amontons
- Lorenzo Romano Amedeo Carlo Avogadro
- Jacques Charles
- Ilya Prigogine
Siehe auch
- System
- Zustandsgleichung (für ideales Gas • für reales Gas) • Ideales Gas • Reales Gas • Van-der-Waals-Radius
- Phase • Phasendiagramm • Tripelpunkt • Isobar • Isotherm • Isochor • Isenthalp • quasi-statisch • Zustandsänderung (adiabatisch • isotherm • isochor • isobar) • Carnot-Prozess • Reversibilität (Physik)
- Statistische Mechanik • Kinetische Gastheorie • Statistische Physik • Maxwell-Boltzmann-Verteilung • Wärmeübertragung • Wärmekapazität
- Thermodynamisches Potenzial • Entropie • Freie Energie • Enthalpie
- Energieerhaltungssatz • Leidenfrost-Effekt • Nernst-Theorem • Wiederkehrsatz • Equilibrierung
- Sankey-Diagramm
- Hess’scher Wärmesatz
Video
- Was ist Entropie? aus der Fernseh-Sendereihe alpha-Centauri
Literatur
Allgemein
- Herbert B. Callen: Thermodynamics and an Introduction to Thermostatistics. 2. Auflage. Wiley Text Books, New York 1985, ISBN 0-471-86256-8
- Constantin Carathéodory: Untersuchungen über die Grundlagen der Thermodynamik In: Mathematische Annalen, 67:355–386, 1909. Carathéodorys Veröffentlichung („Erste axiomatisch strenge Begründung der Thermodynamik“) fand große Beachtung durch Max Planck und Max Born.
- Ulrich Nickel, Lehrbuch der Thermodynamik. Eine verständliche Einführung. 2. Auflage. PhysChem, 2011, ISBN 978-3-937744-06-3
- Max Planck: Vorlesungen über Thermodynamik. eText beim Project Gutenberg
- Karl Stephan, Franz Mayinger: Thermodynamik. Grundlagen und technische Anwendungen. 2 Bände, Springer
- Band 1: Einstoffsysteme. 15. Auflage. 1998, ISBN 3-540-64250-1
- Band 2: Mehrstoffsysteme und chemische Reaktionen. 14. Auflage. 1999, ISBN 3-540-64481-4
- André Thess: Das Entropieprinzip – Thermodynamik für Unzufriedene. Oldenbourg, 2007, ISBN 978-3-486-58428-8
- Gerd Wedler: Lehrbuch der Physikalischen Chemie. Verlag Chemie, Weinheim 1982, ISBN 3-527-25880-9
- Herbert Windisch: Thermodynamik – Ein Lehrbuch für Ingenieure. 4., überarbeitete Auflage. München: Oldenbourg Wissenschaftsverlag 2011, ISBN 978-3-486-70717-5
Chemische Thermodynamik
- Wolfgang Wagner: Chemische Thermodynamik. 4. Auflage. Akademie Verlag, Berlin 1982
- Hans-Heinrich Möbius, Wolfgang Dürselen: Chemische Thermodynamik. 5. Auflage. VEB Verlag für Grundstoffindustrie, Leipzig 1988, ISBN 3-342-00294-8
- Hans-Werner Kammer, Kurt Schwabe: Einführung in die Thermodynamik irreversibler Prozesse. Akademie Verlag, Berlin 1984
- Hans-Joachim Bittrich: Leitfaden der chemischen Thermodynamik. Verlag Chemie, Weinheim 1971, ISBN 3-527-25019-0
- Physikalische Chemie #Allgemeine Lehrbücher
- G. Kortüm: Einführung in die chemische Thermodynamik. 5. Auflage. Verlag Chemie, Weinheim 1966
- Hans Kelker: Angewandte Chemie. Fischer Lexikon, Fischer Taschenbuch, Frankfurt a. M. 1977, ISBN 3-436-02460-0, S. 287–292
Geschichtliches zur Thermodynamik
- Handbuch Der Experimentellen Chemie Sekundarstufe II, Band 7: Chemische Energetik, S. 1–13, Aulis Verlag Deubner, Köln
- Hans Joachim Störig: Kleine Weltgeschichte der Wissenschaft 2. Fischer Taschenbuch, Frankfurt a. M. 1982, S. 88–96; Parkland, Köln 2004, ISBN 3-89340-056-7
Statistische Thermodynamik
Technische Thermodynamik
- Hans D. Baehr, S. Kabelac: Thermodynamik, Grundlagen und technische Anwendungen 13., neu bearb. u. erw. Aufl., Springer Verlag, 2006, ISBN 3-540-32513-1
- Hans D. Baehr, Karl Stephan: Wärme- und Stoffübertragung 5., neu bearb. Aufl., 2006, Springer Verlag, ISBN 3-540-32334-1
- Günter Cerbe, Gernot Wilhelms: Technische Thermodynamik. Theoretische Grundlagen und praktische Anwendungen. 14. Auflage, Hanser Fachbuchverlag, Juni 2005, ISBN 3-446-40281-0
- N. Elsner, A. Dittmann: Grundlagen der Technischen Thermodynamik, Bd. 1 und 2, Akademie Verlag, Berlin 1993
- E. P. Hassel, T. V. Vasiltsova, T. Strenziok: "Einfuehrung in die Technische Thermodynamik"; FVTR GmbH; Rostock 2010; ISBN 978-3-941554-02-3
- Heinz Herwig, Christian H. Kautz: Technische Thermodynamik, Aufl., 2007, Pearson Studium, ISBN 978-3-8273-7234-5
- Dirk Labuhn, Oliver Romberg: Keine Panik vor Thermodynamik!, 1. Auflage, Vieweg, Braunschweig 2005, ISBN 3-8348-0024-4
- Klaus Langeheinecke, Peter Jany, Eugen Sapper: Thermodynamik für Ingenieure. 5. Auflage. Vieweg Verlag, Wiesbaden 2004, ISBN 3-528-44785-0
- Günter Meyer, Erich Schiffner: Technische Thermodynamik, 3. Auflage, VEB Fachbuchverlag, Leipzig 1986
- W. Schneider, S. Haas: Repetitorium Thermodynamik. Oldenbourg Verlag, 2004, 978-3-486-57614-6
- Volker Sperlich: Übungsaufgaben zur Thermodynamik mit Mathcad, Fachbuchverlag Leipzig 2002, ISBN 3-446-21603-0
- Peter Stephan, Karlheinz Schaber, Karl Stephan, und Franz Mayinger Thermodynamik 1. Einstoffsysteme. Grundlagen und technische Anwendungen, Springer Verlag, Berlin, November 1998, 15. Auflage, ISBN 3-540-64250-1
- Wolfgang Wagner: Properties of Water and Steam. / Zustandsgrößen von Wasser und Wasserdampf, Springer Verlag, Berlin, 1. Auflage, August 2002, ISBN 3-540-64339-7
- Klaus Lucas: Thermodynamik. / Die Grundgesetze der Energie- und Stoffumwandlungen, Springer Verlag, Berlin, 5. Auflage, 2006, ISBN 3-540-26265-2
Bemerkungen zu einigen dieser Bücher:hier
Thermodynamik in der Biologie
- Dieter Leuschner: Thermodynamik in der Biologie. Eine Einführung. Akademie Verlag, Berlin 1989, ISBN 3-05-500487-6
Weblinks
 Wikibooks: Thermodynamik – Lern- und Lehrmaterialien
Wikibooks: Thermodynamik – Lern- und Lehrmaterialien- Was ist Entropie? aus der Fernseh-Sendereihe alpha-Centauri
- Eberhard-Karl-Universität Tübingen – 2. Hauptsatz der Wärmelehre, Entropie, Zustandsgleichung realer Gase und Phasenumwandlungen, (Skripte zur Vorlesung Experimentalphysik I Nummer 12; 9-Seitiges PDF)
- Ausführliches Skript zur Thermodynamik und statistischen Physik
- Erklärungen zu den Grundlagen der Thermodynamik (Text und Video)
- Die Hauptsätze der Thermodynamik
- Online-Repetitorium Thermodynamik – mit Videos – (Experimentalphysik) – Universität Würzburg
- Universität Duisburg-Essen, Grundlagen der Technischen Thermodynamik mit Übungsaufgaben und Lösungen
- E-Learning-Kurs zur Thermodynamik
Einzelnachweise und Anmerkungen
- ↑ Wilhelm Gemoll: Griechisch-Deutsches Schul- und Handwörterbuch. München/Wien 1965.
- ↑ Bergmann–Schaefer, Bd. 1, S. 990
- ↑ Handbuch der Experimentellen Chemie Sekundarbereich II, Band 7: Chemische Energetik. Aulis Verlag Deubner, Köln, S. 1
- ↑ Hans Joachim Störig: Kleine Weltgeschichte der Wissenschaften 2, Fischer Taschenbuch, Juni 1982, S. 91
- ↑ Gerd Wedler: Lehrbuch der Physikalischen Chemie. Verlag Weinheim, 1982, S. 59
- ↑ Hans Joachim Störig: Kleine Weltgeschichte der Wissenschaften 2, Fischer Taschenbuch, Juni 1982, S. 93, 1280
- ↑ Lorenz: Abriß der geschichtlichen Entwickelung der Wärmelehre. In: Zeitschrift für Kälteindustrie, 1904, Heft 8, S. 144
- ↑ H. von Helmholtz: Die Thermodynamik chemischer Vorgänge, 1882. In: Wissenschaftliche Abhandlungen von Hermann Helmholtz, Band 2. J. A. Barth, Leipzig 1882, S. 958–978
- ↑ Handbuch der Experimentellen Chemie Sekundarbereich II, Band 7: Chemische Energetik. Aulis Verlag Deubner, Köln, S. 11
- ↑ Handbuch der Experimentellen Chemie Sekundarbereich II, Band 7: Chemische Energetik. Aulis Verlag Deubner, Köln, S. 9
- ↑ Zitat von Max Planck, nach Thieme Chemistry (Hrsg.): Eintrag zu Hauptsätze im Römpp Online. Version 3.14. Georg Thieme Verlag, Stuttgart 2011, abgerufen am 24. August 2011..














Wikimedia Foundation.