- Nucleophile Substitution
-
Die nukleophile Substitution ist ein wichtiger Reaktionstypus in der organischen Chemie. Hierbei reagiert ein Nukleophil in Form einer Lewis-Base (Elektronenpaardonator) mit einer organischen Verbindung vom Typ R–X (R bezeichnet einen Alkyl- oder Arylrest, X ein elektronenziehendes Heteroatom). Das Heteroatom wird dabei durch das Nukleophil ersetzt (siehe Substitution (Chemie)).
In der Anorganischen Chemie ist dieser Typus ebenfalls anzutreffen, ein Beispiel ist die Hydrolyse von Siliciumtetrachlorid.
Inhaltsverzeichnis
Allgemeine Kennzeichen
Nukleophile Substitutionsreaktionen werden meistens in Lösung durchgeführt. Dabei sind die Polarität des Lösungsmittels sowie die Substituenteneinflüsse in den Edukten von entscheidender Bedeutung für die Geschwindigkeit der Reaktion. Ist das Lösungsmittel selbst der nukleophile Reaktionspartner, spricht man von einer Solvolyse.
Edukte
Nukleophile
Als Nukleophile können die verschiedensten Verbindungen eingesetzt werden. Dabei handelt es sich um Anionen oder elektronenreiche Moleküle mit freien Elektronenpaaren (siehe Beispiele unten).
Typ R-X
Das angegriffene Molekül R-X hat eine stark polare Bindung (ungleiche Verteilung der Elektronendichte), z.B C-Cl, C-Br, C-O, C=O oder Si-Cl.
In folgenden Verbindungen kann das Heteroatom beziehungsweise die Heteroatom-haltige Gruppe durch ein Nukleophil substituiert werden:
- Alkylhalogenide: Alkylchloride und Alkylbromide
- Arylhalogenide: Arylchloride und Arylbromide
- Carbonsäurederivate, wie -Chloride, Ester, und -Anhydride
- Sulfonsäureester, z. B. Tosylate (p-Toluolsulfonsäureester) oder Mesylate (Methansulfonsäureester) oder die besonders reaktiven Triflate (Trifluormethansulfonsäureester)
- Oxiran-, Thiiran- und Aziridin-Ringe (→ Heterocyclen)
Mechanismen
Nukleophile Substitutionen werden bei aliphatischen und aromatischen Verbindungen beobachtet: Es gibt aliphatische nukleophile Substitutionen und aromatische nukleophile Substitutionen, wobei erstere wesentlich weiter verbreitet ist.
Darüber hinaus werden die Reaktionen aufgrund der Molekularität in verschiedene Gruppen eingeteilt. Das heißt, die Reaktionen werden danach eingeordnet, wie viele Moleküle am geschwindigkeitsbestimmenden Schritt der Reaktion beteiligt sind. Die beschriebenen Mechanismen SN1 und SN2 sind als Extremfälle der nukleophilen Substitution aufzufassen. Der Übergang dazwischen ist fließend.
Der SNi-Mechanismus ist ein Spezialfall, der gesondert diskutiert wird.
Aromatische nukleophile Substitutionen laufen meistens zweistufig ab, das heißt die Zwischenprodukte sind oft isolierbar (siehe Meisenheimer-Komplexe). Zusätzlich ist ein sogenannter Dehydrobenzol-Mechanismus bekannt, der auch als Arinmechanismus bezeichnet wird.
SN1-Mechanismus
SN1 steht für eine nukleophile Substitution mit einem monomolekularen Mechanismus (ein Molekül ist am geschwindigkeitsbestimmenden Schritt beteiligt. Daher die 1, denn die SN1-Reaktion ist erster Ordnung). Der Reaktionsverlauf ist zweistufig.
Im ersten Schritt wird aus der Verbindung R-X die Gruppe X als Anion freigesetzt. Zurück bleibt ein Carbokation (Carbokation = Carbeniumion) (R+). Danach erfolgt der Angriff des elektronenreichen Nukleophils unter Bildung des Produkts. Die Reaktion ist damit beendet, sie findet bevorzugt in polaren protischen Lösungsmitteln und bei Verbindungen, die relativ stabile Carbokationen bilden (mesomeriestabilisierte oder tertiäre Kohlenstoffatome mit einer positiven Ladung), statt. Außerdem wird der SN1-Mechanismus durch eine relativ kleine Anfangskonzentration der Edukte begünstigt. Das Nucleophil ist nicht am geschwindigkeitsbestimmenden Schritt beteiligt (siehe SN2), stehen aber mehrere Nucleophile zur Verfügung, so findet man im Produkt überwiegend das stärkere Nucleophil wieder.
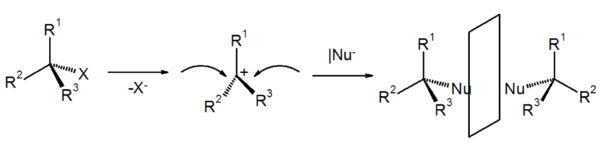
Stereochemie
Der geschwindigkeitsbestimmende Schritt beim SN1-Mechanismus ist die Bildung des idealerweise planaren Carbokations. Die Konfiguration der Ausgangsverbindung wird dadurch aufgehoben.
Theoretisch ist der nachfolgende Angriff des Nukleophils von beiden Seiten gleich wahrscheinlich. Experimentell findet man oft mehr Produkte mit einem Konfigurationswechsel (Ausbeute 50-70%), dies liegt daran, das nach einer Abspaltung der Abgangsgruppe, diese Abgangsgruppe nicht schnell genug durch das Lösungsmittel diffundiert und somit diese Angriffsrichtung versperrt. Die abgehende Gruppe kann sich vor dem nucleophilen Angriff nicht ausreichend entfernen und stellt so für das angreifende Nucleophil eine Behinderung dar. Dies führt manchmal zu einer verstärkten Inversion der Konfiguration. In einem solchen Fall spricht man von einer Teil-Racemisierung.
Beobachtet wurden bei Reaktionen nach SN1 jedoch alle stereochemischen Möglichkeiten von vollständiger Inversion bis zur Racemisierung, wobei die Bildung eines Racemates die Regel ist. Ein racemisches Produkt wäre die Folge, da der Angriff von der der austretenden Gruppe gegenüberliegenden Seite einen Konfigurationswechsel (=Inversion), der von derselben Seite die Erhaltung der Konfiguration (Retention) zur Folge hätte.
SN2-Mechanismus
Eine nukleophile Substitution mit einem bimolekularen Mechanismus (Reaktion ist 2. Ordnung) wird kurz mit SN2 bezeichnet und verläuft einstufig. Der nukleophile Angreifer (auch "Angriff der Nukleophilen") nähert sich dem positiven Kern und es bildet sich eine trigonale Bipyramide mit schwachgebundenen axialen Liganden. Es folgt Inversion (bei chiralen Molekülen: Walden-Umkehr; auch "Regenschirmprinzip nach Krieger" genannt).

In dem Maße, wie sich das Nukleophil dem Kern nähert, entfernt sich das abgehende Teilchen oder die substituierte Gruppe bis die Abgangsgruppe das Molekül verlässt.
Diese Substitution findet bevorzugt in polaren aprotischen Lösungsmitteln und an primären Kohlenstoffatomen statt (da es bei tertiären Kohlenstoffatomen zu einer sterischen Hinderung kommt). Außerdem wird der SN2-Mechanismus durch eine relativ große Anfangskonzentration der Edukte begünstigt.SN2-Mechanismen am ungesättigten Kohlenstoffatom
Betrachtet man chlorsubstituierte ungesättigte Verbindungen, wie Vinylchlorid (C2H3Cl) oder Chlorbenzol (C6H5Cl), so wird gefunden, dass diese ungesättigten Verbindungen nur äußerst schlecht von Nucleophilen wie dem Hydroxid-Ion oder dem Amid-Ion angegriffen werden. Alkylhalogenide, also die gesättigten Halogenverbindungen, reagieren meist bereits bei Raumtemperatur, während bei der Reaktion von Chlorbenzol mit Hydroxid-Ionen Temperaturen von 200°C nötig sind. Verantwortlich für dieses reaktionsträge Verhalten ist die erhöhte Elektronendichte an den ungesättigten Kohlenstoffatomen. Dadurch wird der Angriff eines Nucleophils erschwert; ungesättigte Kohlenstoffatome ziehen das gemeinsame Elektronenpaar der zu substituierenden Gruppe (z.B. die C-Cl - Bindung im Vinylchlorid oder im Chlorbenzol) stärker zu sich, was das Abstrahieren des Chloratoms erschwert.
Die Einführung elektronenziehender Gruppen im Benzol und seinen substituierten Derivaten, führte zur Entdeckung eines neuen Reaktionswegs, den man als SN2 (aromatisch) bezeichnet. Betrachtet man Chlorbenzol und vergleicht die Reaktionsgeschwindigkeit bei einem nucleophilen Angriff mit der Reaktion von p-Nitrochlorbenzol, so stellt man eine beträchtliche Steigerung der Umsatzgeschwindigkeit fest. Der genaue Mechanismus wird unter dem Artikel Nukleophile aromatische Substitution beschrieben.
SNi-Mechanismus
Die Gewinnung von Alkylchloriden durch nukleophile Substitution von Alkanolen mit Thionylchlorid erfolgt nach einem sogenannten SNi-Mechanismus. Aus einem enantiomerenreinen Alkanol als Edukt wird ein Alkylchlorid mit gleicher Konfiguration erhalten. Die SNi-Reaktion verläuft also unter Retention (Erhalt der Konfiguration).
Nachbargruppenbeteiligung
Nucleophile Substitutionen können auch durch molekülinterne Prozesse gesteuert werden. So kann es zu einer Beteiligung, der schon am betrachteten Kohlenwasserstoff gebundenen Substituenten kommen. Diese intramolekulare Reaktion ist bevorzugt, da die Wahrscheinlichkeit hoch ist, mit den am benachbarten C-Atom liegenden Substituenten zusammenzustoßen (Dieses Nucleophil kann nicht durch zum Beispiel das Lösungsmittel vom Substrat entfernt werden).
Hierbei fungiert die Nachbargruppe (Substituent) als Nucleophil, welches über einen Rückseitenangriff die Abgangsgruppe abspalten lässt. Es bildet sich übergangsweise ein zyklisches System. Ein solcher Zyklus kann einerseits durch eine hohe Ringspannung (kleine Ringe) oder andererseits durch einen Angriff eines externen Nucleophils geöffnet werden. Im zweiten Fall wird demnach unter zweifacher Inversion das Retentionsprodukt erhalten.
Beispiele
Substitution am Alkylkohlenstoff/Arylkohlenstoff
Sauerstoff als Nukleophil
- Alkylchloride reagieren mit Hydroxid-Ionen zu Alkoholen unter Freisetzung von Chlorid-Ionen. Analog dazu reagieren chlorierte Aromaten zu Phenolen:

- Alkylchloride reagieren mit Wasser zu protonierten Alkoholen und Chlorid (Hydrolyse):

- Aliphatische Ether und Phenolether können durch nukleophile Substitution von Chlorid durch Alkoholate an Alkyl- oder Arylchloriden gewonnen werden. Diese Reaktion wird auch als Williamsonsche Ethersynthese bezeichnet.

- Die Synthese von Estern erfolgt durch die Substitution von Chlorid durch Carbonsäuren:

- Arylchloride reagieren mit Cyanat zu Arylcyanaten und Chlorid:

- Aromatische Sulfonsäuren reagieren in Alkalischmelzen zu Phenolen und Sulfit.
Stickstoff als Nukleophil
- Aliphatische primäre Amine entstehen durch den Austausch des Halogenids gegen die Aminogruppe (-NH2). Diese Reaktion findet in Ammoniak als Lösungsmittel statt und wird auch als Ammonolyse bezeichnet.
- Zur Gewinnung sekundärer Amine wird die Reaktion nicht in Ammoniak sondern mit einem weiteren Amin als Lösungsmittel durchgeführt (→ Aminolyse).
- Tertiäre Amine entstehen durch die Umsetzung mit einem sekundären Amin,
- Tetraalkylammoniumsalze durch die Umsetzung mit einem tertiären Amin.
- Als Gabriel-Synthese wird die Reaktion bezeichnet, bei der ein Alkylchlorid oder -bromid mit Phthalimid umgesetzt wird:

Schwefel als Nukleophil
- Die Reaktionen von Alkyl- und Arylhalogeniden mit Hydrogensulfid und Thiolaten führen analog zu denen mit den Sauerstoff-Homologen Hydroxid und Alkoholaten zu Thiolen und Thioethern.
- Mit Thioharnstoff reagieren Alkylhalogenide zu Isothiuronium-Salzen.
- Durch Substitution des Halogens mit Hydrogensulfit entstehen Sulfonsäuren.
Halogenide als Nukleophil
- Werden Alkyl- oder Arylchloride beziehungsweise -bromide mit einem Überschuss an Fluorid (in polaren, aprotischen Lösungsmitteln) oder Iodid (in Aceton) umgesetzt, entstehen aliphatische oder aromatische Fluoride oder Iodide. Die Reaktion mit Iodid wird als Finkelstein-Reaktion bezeichnet.
Phosphor als Nukleophil
- Alkylchloride reagieren mit Alkyl- oder Arylphosphanen zum entsprechenden Phosphoniumsalz. Aus organischen Phosphoniumsalzen werden die Olefinierungsreagenzien für die Wittig-Reaktion gewonnen.

Hydrid als Nukleophil
- Alkane können durch Reaktion von Alkylhalogeniden mit Hydrid als Substituent hergestellt werden. Hydrid-Donator ist Lithiumaluminiumhydrid.

Siehe auch
Reaktionsmechanismen der Organischen ChemieCycloaddition | Elektrophile Addition | Elektrophile Substitution | Eliminierung | Nukleophile Addition | Nukleophile Substitution | Oxidation | Radikalische Addition | Radikalische Substitution | Reduktion | Umlagerung






Wikimedia Foundation.