- Entropie (Thermodynamik)
-
Dieser Artikel wurde den Mitarbeitern der Redaktion Physik zur Qualitätssicherung aufgetragen. Wenn Du Dich mit dem Thema auskennst, bist Du herzlich eingeladen, Dich an der Prüfung und möglichen Verbesserung des Artikels zu beteiligen. Der Meinungsaustausch darüber findet derzeit nicht auf der Artikeldiskussionsseite, sondern auf der Qualitätssicherungs-Seite der Physik statt.

Physikalische Größe Name Entropie Formelzeichen der Größe S Größen- und
Einheiten-
systemEinheit Dimension SI 
L2·M·T−2·θ−1 Die Entropie (griechisches Kunstwort εντροπία [entropía], von εν~ [en~] – ein~, in~ und τροπή [tropē] – Wendung, Umwandlung) ist eine fundamentale thermodynamische Zustandsgröße. Sie beschreibt die Zahl der Mikrozustände, durch die der beobachtete Makrozustand des Systems realisiert werden kann. Sie ist eine extensive Zustandsgröße, jedem Zustand eines thermodynamischen Systems kann also ein Wert der Entropie zugeordnet werden. Die statistische Physik interpretiert diese Zahl als Maß für das vom System erreichbare Phasenraumvolumen, in der klassischen Thermodynamik sind Gleichgewichtszustände mit gleicher Entropie adiabatisch äquivalent.
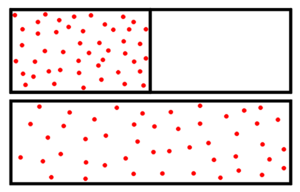 Dem Gas steht nach dem Entfernen der Zwischenwand ein größerer Raum zur Verfügung. Es existieren nach der Expansion also mehr Mikrozustände und das System besitzt eine höhere Entropie.
Dem Gas steht nach dem Entfernen der Zwischenwand ein größerer Raum zur Verfügung. Es existieren nach der Expansion also mehr Mikrozustände und das System besitzt eine höhere Entropie.
Einordnung in die Thermodynamik
Die klassische Thermodynamik ist ein Denkmodell, das die energetischen Wechselwirkungen des Systems mit seiner Umgebung beschreibt. Das System hat dabei zwei prinzipielle Möglichkeiten mit seiner Umgebung Energie auszutauschen, in Form von Wärmeübertragung und in Form von Arbeit. Im Zuge eines solchen Energieaustausches ändert sich die Entropie des Systems und der Umgebung. Nur wenn die Summe dieser beiden Entropieänderungen positiv ist, das System nach der Änderung also mehr Mikrozustände besitzt als vorher, erfolgt die Änderung spontan.
Grundlagen
Die Entropie S (Einheit J/K) ist eine extensive Zustandsgröße eines physikalischen Systems und verhält sich bei Vereinigung mehrerer Systeme additiv wie auch das Volumen, die Elektrische Ladung oder die Stoffmenge. Dividiert man durch die Masse des Systems, erhält man die spezifische Entropie s mit der Einheit J/(kg·K) als intensive Zustandsgröße. Der deutsche Physiker Rudolf Clausius führte diesen Begriff 1865 zur Beschreibung von Kreisprozessen ein.
Das Differential dS (ein nicht-kursives d wird benutzt, um herauszuheben, dass es sich um ein vollständiges Differential, das heißt eine Zustandsgröße, handelt) ist nach Clausius bei reversiblen Vorgängen das Verhältnis von übertragener Wärme δQ und absoluter Temperatur T:
Diese Entropieänderung ist bei Wärmezufuhr positiv, bei Wärmeabfuhr negativ. Clausius beschrieb auch die Entropievermehrung ohne Wärmeübertragung durch irreversible Vorgänge in einem isolierten System mit der Ungleichung:
Man unterscheidet heute zwischen der transportierten Entropie (1. Gleichung) und der produzierten Entropie (2. Gleichung) und fasst − allgemein gültig, also auch für das nicht adiabate System – beide Gleichungen zusammen:
Dabei ist δWdiss die innerhalb des Systems dissipierte Arbeit, die immer positiv ist, also dem System zugeführt wird (z. B. Reibungsarbeit). Die 3. Gleichung ist eine Form des zweiten Hauptsatzes der Thermodynamik. Das Differential dS existiert allerdings nur für quasi-statische Zustandsänderungen, d. h., dass eine Folge von Zuständen messtechnisch erfasst werden kann (Zustände mit nur geringen Ungleichgewichten). Bei dem im Bild dargestellten Vorgang im adiabaten System, bei dem nur Anfangszustand und Endzustand angegeben werden können, wäre das nicht der Fall. Für ein ideales Gas kann aber die Entropiedifferenz auf einfache Weise über einen reversiblen isothermen Ersatzprozess (Abgabe von Arbeit und Aufnahme von Wärme aus der Umgebung) berechnet werden, wie dies im Abschnitt Beispiele geschehen ist.
Geschichte des Begriffes „Entropie“
 Schmelzendes Eis in einem warmen Raum ist ein einfaches Beispiel für die Erhöhung der Entropie (Erstmals beschrieben 1862 von Rudolf Clausius).
Schmelzendes Eis in einem warmen Raum ist ein einfaches Beispiel für die Erhöhung der Entropie (Erstmals beschrieben 1862 von Rudolf Clausius).Die Entropie ist neben der Energie der wichtigste Begriff der Thermodynamik, und es ist hilfreich, sich für ein besseres Verständnis an den Ausgangspunkt dieser Wissenschaft zu begeben und die Entwicklung zu rekapitulieren. Im Jahre 1712 wurde die erste Dampfmaschine von Thomas Newcomen in einem Bergwerk installiert, um Wasser zu pumpen. Die Maschine wurde ihrer Aufgabe gerecht, benötigte aber sehr viel Brennstoff. Zu diesem Zeitpunkt war der Zusammenhang zwischen Energie und Wärme völlig unklar und es sollten noch über 130 Jahre vergehen, bis Julius Mayer den 1. Hauptsatz der Thermodynamik publizierte. Ab 1764 verbesserte James Watt die Dampfmaschine und konnte deren Wirkungsgrad auf über 1% mehr als verdoppeln, und das ohne Kenntnis der formalen thermodynamischen Gesetze. Erst 60 Jahre später hatte der junge französische Ingenieur Sadi Carnot die entscheidende Idee, die er 1824 publizierte. Inspiriert von der Arbeit seines Vaters über Wassermühlen beschrieb Carnot eine Dampfmaschine durch einen zyklischen Prozess, bei dem Wärme von einer heißen Quelle zu einer kalten Senke fließt und dabei Arbeit leistet. Das Verhältnis von entnommener mechanischer Arbeit ΔW zu eingeleiteter Wärme ΔQ war der Wirkungsgrad η:
In seiner ursprünglichen Schrift vertrat Carnot die Meinung, dass Wärme eine Art unwägbarer Stoff sei, der immer von einem heißen zu einem kühleren Körper fließe, ähnlich wie Wasser sich immer bergab bewegt. Und genau wie herabstürzendes Wasser könne Wärme umso mehr Arbeit leisten, je höher das Gefälle sei, insbesondere könne die Maschine nicht mehr Arbeit leisten als Wärme zugeführt wurde. Carnot korrigierte sich später und erkannte bereits ein Jahrzehnt vor Mayer, Joule und Thomson die Äquivalenz von Wärme und Energie. Er war also seiner Zeit weit voraus, starb jedoch jung und sein Werk blieb zunächst unbemerkt. Erst Clausius formulierte den Zusammenhang von Temperaturdifferenz – der Quelle und Senke – mit dem Wirkungsgrad der Carnot-Maschine und dass dieser Wirkungsgrad nicht von einer anderen Wärmekraftmaschine überschritten werden kann, da sonst Wärme spontan von einem kalten zu einem heißen Körper fließen würde. Die Unmöglichkeit eines solchen Vorgangs in der Natur bezeichnet man heute als 2. Hauptsatz der Thermodynamik. Clausius formulierte ihn mit einem Kreisprozess:
- Es existiert keine zyklisch arbeitende Maschine, deren einzige Wirkung Wärmetransport von einem kühleren zu einem wärmeren Reservoir ist.
Einfacher ausgedrückt besagt also der 2. Hauptsatz, dass Temperaturdifferenzen sich in der Natur nicht spontan vergrößern können. Clausius konnte mit dieser Forderung den Satz
für beliebige Kreisprozesse herleiten. Das Gleichheitszeichen gilt dabei nur für reversible Prozesse. Mit diesem Satz von Clausius liegt es auf der Hand, die Größe
differentiell zu definieren. Rudolf Clausius nannte diese Größe Entropie, ein Kunstwort, welches dem Begriff der Energie nachempfunden wurde und in etwa mit Wandlungsgehalt zu übersetzen ist – im Gegensatz zum Wärmeinhalt. Es wurde mit der Zeit üblich, den 2. Hauptsatz direkt mit der Entropie zu formulieren, was keineswegs zu einem tieferen Verständnis führt. Erst Jahrzehnte später konnte Ludwig Boltzmann mit seiner statistischen Mechanik eine Erklärung für die Entropie als Maß für die erreichbaren Mikrozustände des Systems finden. Wärme ist zufällig über Atome und Moleküle verteilte Energie und fließt von heiß nach kalt, weil der umgekehrte Weg zu unwahrscheinlich ist.
Im Jahre 1999 haben die theoretischen Physiker Elliott Lieb und Jakob Yngvason die Definition der Entropie in der phänomenologischen Thermodynamik auf eine streng axiomatische Basis gestellt. Diese Definition macht keinen Gebrauch von Größen wie „Wärme“ und „Temperatur“, die sich ohne Entropie nicht exakt definieren lassen, sondern beruht auf dem Konzept der adiabatischen Erreichbarkeit.
Hilfestellung zum Verständnis
Energie wird – entgegen der landläufigen Redeweise – im physikalischen Sinn nicht verbraucht, sondern nur umgewandelt, z. B. in mechanische Arbeit und Wärme (1. Hauptsatz der Thermodynamik – Energieerhaltung). Einem Benzinmotor wird also im Laufe eines Zyklus dieselbe chemische Energiemenge in Form von Kraftstoff zugeführt, wie als Antriebsarbeit und Wärme abgeführt wird. Da auch die Antriebsarbeit durch Reibung schließlich in Wärme umgesetzt wird, landet am Ende der gesamte Energieinhalt des Kraftstoffes als Wärmemenge in der Umgebung, sieht man von eventuell in potenzielle Energie oder mechanisch in Deformationsenergie umgewandelten Anteilen ab. Die Energie wurde also nicht verbraucht, sondern lediglich umgewandelt; es ist sinnvoll, von einer Energieentwertung zu sprechen. Man benötigte also eine Größe, um die Arbeitsfähigkeit der Energie zu beschreiben, da die Energiemenge alleine nichts über die Arbeitsfähigkeit aussagt. So enthalten die Weltmeere eine riesige Energiemenge. Da diese aber bei Umgebungstemperatur vorliegt, kann damit keine Arbeit verrichtet werden. Deshalb erscheint es wegen Gleichung (1) konsequent, die gewichtete Entropiedifferenz
 , die z. B. auch bei Differenzen der sog. Freien Energie F = U-TS auftritt (U = Innere Energie), als „Abwärme“, „Verlust-Energie“ oder ähnlich zu bezeichnen, wie es z. B. in der Physikdidaktik häufig geschieht.
, die z. B. auch bei Differenzen der sog. Freien Energie F = U-TS auftritt (U = Innere Energie), als „Abwärme“, „Verlust-Energie“ oder ähnlich zu bezeichnen, wie es z. B. in der Physikdidaktik häufig geschieht.Clausius fand heraus, dass man mit einem Kreisprozess eine gegebene Wärmemenge umso besser in Arbeit umwandeln kann, je höher die Temperatur ist, bei der sie der Maschine zugeführt wird (siehe Carnot-Wirkungsgrad). Am Beispiel des Motors wird die chemische Energie des Kraftstoffs durch die Verbrennung dem Motor bei ca. 2000–2500 °C zugeführt und verlässt ihn wieder zu je etwa einem Drittel bei ca. 800 °C durch die Abgase, bei ca. 50 °C durch den Kühler sowie über die Räder. Mit Hilfe der Gleichungen von Clausius kann man nun vorhersagen, wie viel der Kreisprozess des Motors maximal erbringen könnte. Die zugeführte Energie hat dabei eine geringe Entropie, während die Abwärme eine hohe Entropie hat. Aus der Differenz lässt sich die mögliche Arbeit berechnen. Die Aussage des sog. 2. Hauptsatzes der Thermodynamik, dass die Entropie bei einem beliebigen reversiblen Kreisprozess konstant ist, während sie bei irreversiblen Kreisprozessen zunehmen muss, ist zu folgender Aussage äquivalent:
Bei isothermen Prozessen (T = konstant), bei denen die Freie Energie zunimmt (ΔF > 0), kann maximal die Arbeit δA = ΔF gewonnen werden. (Man beachte, dass hier nicht ΔU steht, sondern ΔF).
Problematik des Begriffs Entropie
In populärwissenschaftlichen Büchern, aber auch in vielen Lehrbüchern wird die Entropie mit Unordnung gleichgesetzt. Diese Analogie trifft für einige Systeme zu, z. B. besitzt ein geordneter Kristall eine viel geringere Entropie als seine Schmelze. Für andere Systeme ist diese Betrachtung eher problematisch, z. B. besitzt eine geordnete Biomembran in Wasser eine höhere Entropie als ihre ungeordneten, in Wasser gelösten Bestandteile (siehe Beispiele unten). Das Problem besteht in erster Linie darin, dass der umgangssprachliche Begriff Unordnung nicht eindeutig definiert ist und die Entropie kein Maß für die Symmetrie des Systems darstellt, sondern für die Anzahl der mikroskopisch erreichbaren Zustände, unabhängig von ihrem wie auch immer definierten Ordnungsgrad. Insbesondere in Lehrbüchern der theoretischen Physik wird der Begriff Unordnung deshalb gemieden.
Verwirrung entsteht auch dadurch, dass der Begriff der Entropie in unterschiedlichen Disziplinen mit Bezug auf unterschiedliche Phänomene verwendet wird. Die Entdeckung der Entropie im Zusammenhang mit der Thermodynamik und ihre zentrale Rolle für diese Theorie verhinderte nicht ihre Übertragung auf andere Bereiche, wie z. B. die Informationstheorie. Die Entropie ist eine statistisch definierte Größe und kann in vielen Kontexten sinnvoll verwendet werden. Unbeschadet dessen können die Definitionen in den Einzeldisziplinen sogar widersprüchlich sein. So nutzte Norbert Wiener den Begriff der Entropie ebenso zur Beschreibung von Informationsphänomenen wie Claude Elwood Shannon, allerdings mit einem negativen Vorzeichen. Dass sich die Definition von Shannon durchgesetzt hat, ist vor allem der besseren technischen Verwertbarkeit seiner Arbeiten zuzuschreiben. Es wird aber aus diesem Beispiel deutlich, dass bei einer interdisziplinären Anwendung des Entropiebegriffes mindestens Vorsicht und eine genaue Quellenanalyse geboten ist.[1]
Die Entropie ist keine direkt messbare statistische Größe, wie z. B. die Temperatur und der Druck. Es können nur Änderungen der Entropie erfasst werden, und sie ist auch keine strenge Erhaltungsgröße wie Energie, Masse, Teilchenzahl oder Ladung eines Systems. Dies ist auch ein wesentlicher Unterschied zwischen erstem und zweitem Hauptsatz der Thermodynamik. Während der erste Hauptsatz nichts anderes als die Formulierung des streng gültigen Energieerhaltungssatzes in der Sprache der Thermodynamik ist, stellt der zweite Hauptsatz nur eine statistisch gerechtfertigte Behauptung dar. Allerdings ist die Wahrscheinlichkeit für einen Verstoß gegen den zweiten Hauptsatz in makroskopischen Systemen extrem gering. Er kann allerdings nicht direkt aus den mikroskopischen Gleichungen gefolgert werden und wurde sogar im Rahmen der klassischen Mechanik durch Poincaré widerlegt. All diese Eigenschaften führen zu Problemen beim Verständnis des Begriffs der Entropie.
Entropie in der Thermodynamik
Ein idealer, jederzeit umkehrbarer Prozess ohne Reibungsverluste wird auch reversibel genannt. Oft bleibt die Entropie während eines Prozesses unverändert, ΔS = 0, bekanntes Beispiel ist die adiabate Kompression und Expansion im Zyklus einer Carnot-Maschine. Man nennt Zustandsänderungen mit konstanter Entropie auch isentrop, allerdings sind nicht alle isentropen Zustandsänderungen adiabatisch. Ist ein Prozess adiabatisch und reversibel folgt jedoch stets, dass er auch isentrop ist.
Wird in einem Kreisprozess bei der Temperatur Th die Wärme Qh aufgenommen und die Wärmemenge Ql bei Tl wieder abgegeben, gilt, dass sich die Entropie nicht ändert:
 ; oder
; oder 
sofern Wärmeaufnahme und Abgabe reversibel erfolgen.
Daraus lassen sich die maximale Energieleistung A = Qh − Ql und der maximale Wirkungsgrad ableiten.
 Mischungsentropie
MischungsentropieDas Bild rechts zeigt die Mischung einer braunen Farbe in Wasser. Zu Beginn ist die Farbe ungleichmäßig verteilt. Nach längerem Warten nimmt das Wasser eine gleichmäßige Färbung an.
Die Entropie ist ein Maß für Unwissenheit. Als Maß für Unordnung muss man genau auf die Begrifflichkeit achten. So ist im Bildbeispiel die Flüssigkeit im rechten Glas zwar „ordentlicher“ verrührt, aber durch die große Durchmischung von Wasser- und Farbteilchen herrscht dort eine größere Unordnung. Mithin ist dort die Entropie höher als im linken Glas. Von der Farbe wissen wir, dass sie im rechten Glas überall im Wasser verteilt ist. Das linke Bild sagt uns mehr. Wir können Bereiche ausmachen, in denen Farbe in hoher Konzentration anzutreffen ist, oder Bereiche, die frei sind von Farbe.
Die Mischungsentropie lässt sich berechnen. Josiah Willard Gibbs wies auf den Widerspruch hin, dass der Entropiezuwachs auch auftreten sollte, wenn statt der Tinte Wasser ins Wasserglas gegossen wird (Gibbssches Paradoxon).
Die Zahl der Anordnungen der Farbmoleküle am Anfang ist deutlich geringer als die, wenn sich die Farbe im gesamten Volumen verteilen kann. Denn die Farbmoleküle sind nur auf wenige Bereiche konzentriert. Im rechten Bild können sie sich im gesamten Glas aufhalten. Die Entropie ist hier größer, weshalb das System im Lauf der Zeit dieser Gleichverteilung zustrebt.
Die Entropie bleibt nur dann unverändert, wenn die Prozesse reversibel verlaufen. Reale Zustandsänderungen sind immer mit Energieverlusten (z. B. durch Reibung) verbunden, wodurch sich die Entropie erhöht. Eine Verringerung der Gesamtentropie in einem geschlossenen System ist nicht möglich. Aber die Entropie kann lokal verkleinert werden, wenn sie an anderen Orten des Systems entsprechend anwächst.
Siehe auch: Mischungsentropie eines idealen Gasgemischs
Zweiter und dritter Hauptsatz
Rudolf Julius Emanuel Clausius hatte erkannt, dass die durch
differentiell gegebene Größe eine extensive Zustandsgröße darstellt, also unabhängig vom Reaktionspfad und proportional zur Systemgröße ist. Die Bezeichnung δQreversibel statt dQ betont, dass die vom System aufgenommene oder abgegebene Wärme wegabhängig (Beispiel siehe Kreisprozess) und deshalb nur bei reversibler Prozeßführung ein vollständiges Differential ist.
Clausius fand außerdem, dass in einem isolierten System die Entropie monoton wächst:
Er formulierte diese Beobachtung im 2. Hauptsatz der Thermodynamik als Negation der Existenz eines Perpetuum mobile zweiter Art:
„Es existiert kein Kreisprozess, dessen einzige Wirkung darin besteht, Wärme von einem kälteren Reservoir zu einem wärmeren Reservoir zu transportieren.“
Offenbar hätte man sonst eine unerschöpfliche Energiequelle konstruiert. Äquivalent dazu ist die Formulierung von William Thomson:
„Es existiert kein Kreisprozess, der eine Wärmemenge aus einem Reservoir entnimmt und vollständig in Arbeit verwandelt.“
Im Gegensatz zu den bereits bekannten extensiven Größen von thermodynamischen Systemen, wie Energie E, Volumen V und Masse m, entzog sich die Entropie zunächst dem tieferen Verständnis. Die Entropie konnte erst im Rahmen der statistischen Mechanik von Ludwig Boltzmann befriedigend als Maß für das Phasenraumvolumen erklärt werden, das von der Phasentrajektorie des Systems unter Einhaltung der Konstanz ausgewählter makroskopischer Observablen, wie Temperatur T, Volumen V oder Teilchenzahl N, erreicht werden kann.
Anschaulich ist die Entropie demnach ein Maß für fehlende Information über den tatsächlichen Mikrozustand, wenn lediglich eine geringe Anzahl beobachtbarer Größen zur Charakterisierung des Makrozustands vorliegen. Die Ergodenhypothese behauptet, dass die Trajektorie des Systems tatsächlich im Laufe der Zeit das gesamte durch die Entropie gemessene Phasenvolumen überdeckt. Systeme, die dieses Verhalten zeigen, nennt man auch ergodisch. Nur bei diesen kann der 2. Hauptsatz sinnvoll angewandt werden. Eng damit verbunden ist die Irreversibilität von Prozessen in der Natur.
Der dritte Hauptsatz (der so genannte „Nernstsche Wärmesatz“) legt die Entropie einer perfekt kristallinen Substanz, bei der beispielsweise keine Spinentartung auftritt, am absoluten Nullpunkt als Null fest:
Eine Folgerung ist beispielsweise, dass die Wärmekapazität eines Systems bei tiefen Temperaturen verschwindet, und vor allem, dass der absolute Temperaturnullpunkt nicht erreichbar ist (das gilt auch bei Spinentartung).
Beispiele
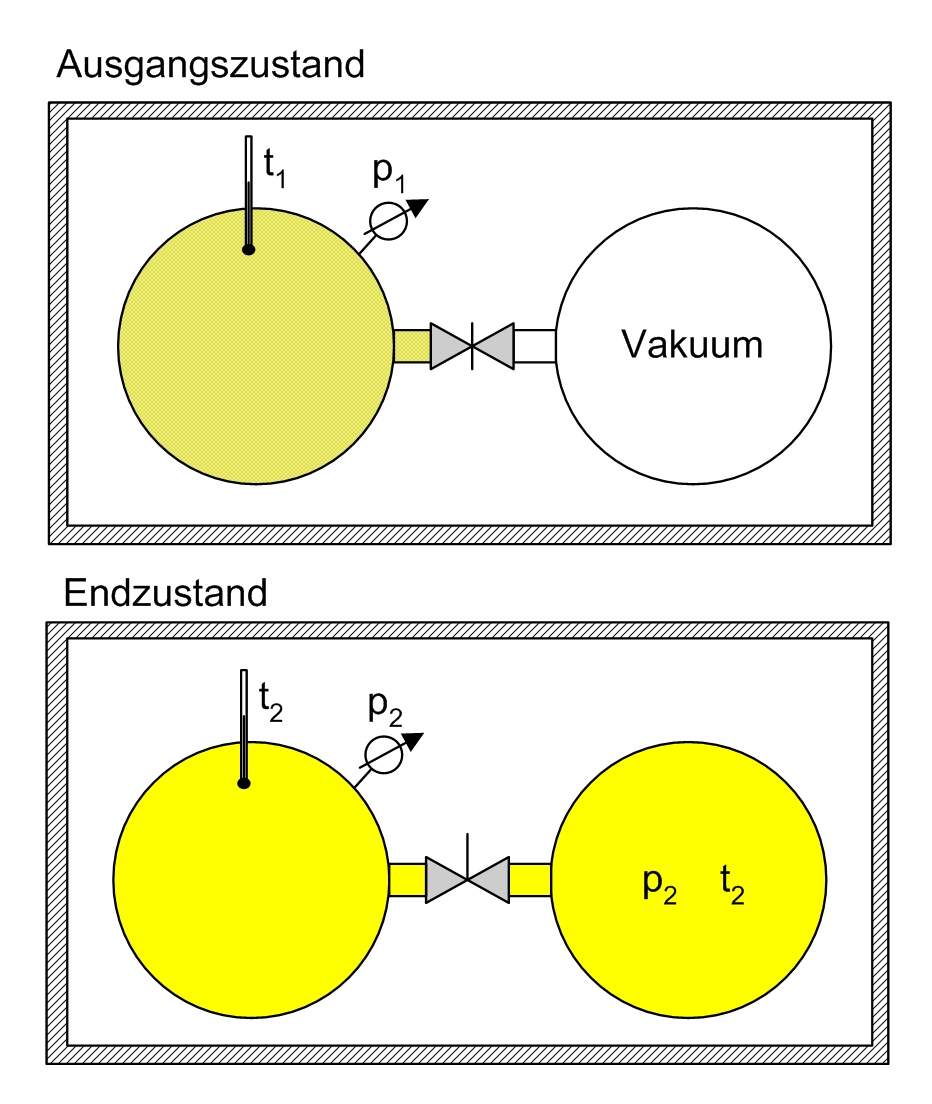
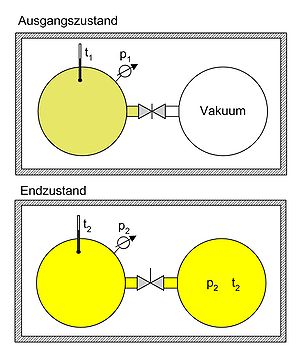 Überströmversuch von Gay Lussac. Der Versuch mit einem idealen Gas in einem abgeschlossenen System zeigt, dass sich nach dem Druck- und Temperaturausgleich die Anfangstemperatur einstellt (t2 = t1). Da sich die innere Energie nicht ändern konnte, kann im Umkehrschluss gefolgert werden, dass die innere Energie einer bestimmten Menge des idealen Gases nur temperaturabhängig, nicht aber druck- oder volumenabhängig ist.
Überströmversuch von Gay Lussac. Der Versuch mit einem idealen Gas in einem abgeschlossenen System zeigt, dass sich nach dem Druck- und Temperaturausgleich die Anfangstemperatur einstellt (t2 = t1). Da sich die innere Energie nicht ändern konnte, kann im Umkehrschluss gefolgert werden, dass die innere Energie einer bestimmten Menge des idealen Gases nur temperaturabhängig, nicht aber druck- oder volumenabhängig ist.- Expansionsversuch von Gay-Lussac
In der Einleitung wird das Experiment von Gay-Lussac beschrieben. Wie groß ist nun die Entropieänderung in dem beschriebenen Versuch? Da die Entropie eine Zustandsgröße ist, ist sie wegunabhängig. Anstatt die Trennwand herauszuziehen, kann man sie auch langsam nach rechts schieben, bis das Endvolumen erreicht ist. Für eine infinitesimale Verschiebung vergrößert sich das Volumen um dV, die Entropie steigt um dS = δQ / T. Aus dem ersten Hauptsatz dU = δQ + δW folgt mit dU = 0 und δW = − pdV, da ausschließlich Volumenarbeit verrichtet wird:
Aus der Zustandsgleichung für ideale Gase (N ist die Anzahl der Gasatome):
folgt:
 .
.
Hieraus ergibt sich durch Integration sofort:
Da im obigen Beispiel N = 47 Atome eingezeichnet sind, gilt:
 .
.
Realistischer wäre z. B. 1 mol Atome, also
 Atome, womit sich
Atome, womit sichergibt.
- Entropiezunahme bei Expansion, statistisch
Die Entropie eines Makrozustands lässt sich auch über sein statistisches Gewicht W (die Anzahl seiner Mikrozustände) ermitteln. Sind n Moleküle auf zwei Raumhälften so verteilt, dass sich in der einen Hälfte n1 und in der anderen n2 Moleküle befinden, dann ist das statistische Gewicht
 und die Entropie dieses Zustands
und die Entropie dieses Zustands  .
.
Befindet sich ein ganzes Mol (n=NA) in einer Hälfte (und in der anderen nichts), dann ist
 und die Entropie
und die Entropie  . Bei gleichmäßiger Aufteilung wird
. Bei gleichmäßiger Aufteilung wird
 .
.
Die Fakultät kann man mit der Stirling-Formel annähern, wobei man sich auf
 beschränken kann. Der Logarithmus von nn ist
beschränken kann. Der Logarithmus von nn ist  . Damit wird
. Damit wirdund
![\ln\left[W\left(\frac{N_A}{2},\frac{N_A}{2}\right)\right]\approx6\cdot10^{23}\cdot\ln(6\cdot10^{23})- 2\cdot3\cdot10^{23}\cdot\ln(3\cdot10^{23})=6\cdot10^{23}\cdot(\ln6-\ln3)](c/78c4e4245cb10b39193fc165b678226d.png) .
.
Mit kB = 1,3807·10-23 J/K und NA = 6,0220·1023 Mol-1 erhält man für die Entropie nach der Expansion
![S=k_B\cdot\ln\left[W\left(\frac{N_A}{2},\frac{N_A}{2}\right)\right]=5{,}76\;\mbox{J/K}](b/80bc5f191a70c40d70b44e37836bd7fa.png) , wie aus der thermodynamischen Rechnung.
, wie aus der thermodynamischen Rechnung.
- Zahlenbeispiel
In einem System, welches mit seiner Umgebung weder Masse noch Energie austauscht, kann die Entropie niemals spontan abnehmen. Beispiel: Ein Kilogramm Wasser besitzt bei 10 °C die Entropie
 , bei 20 °C
, bei 20 °C  , bei 30 °C
, bei 30 °C  . 1 kg kaltes Wasser (10 °C) und 1 kg warmes Wasser (30 °C) können bei Berührung spontan in den Zustand 2 kg lauwarmes Wasser (20 °C) übergehen, weil die Entropie des Anfangszustandes (151 + 437 = 588) kleiner ist als die Entropie des Endzustandes (297 + 297 = 594). Die spontane Umkehrung dieses Vorganges ist nicht möglich, weil sich hierbei die Entropie des aus 2 kg Wasser bestehenden Systems von 594 J/K auf 588 J/K verringern müsste, was dem zweiten Hauptsatz der Thermodynamik widerspräche.
. 1 kg kaltes Wasser (10 °C) und 1 kg warmes Wasser (30 °C) können bei Berührung spontan in den Zustand 2 kg lauwarmes Wasser (20 °C) übergehen, weil die Entropie des Anfangszustandes (151 + 437 = 588) kleiner ist als die Entropie des Endzustandes (297 + 297 = 594). Die spontane Umkehrung dieses Vorganges ist nicht möglich, weil sich hierbei die Entropie des aus 2 kg Wasser bestehenden Systems von 594 J/K auf 588 J/K verringern müsste, was dem zweiten Hauptsatz der Thermodynamik widerspräche.- Biomembranen
Gibt man Lipide, bei Lebewesen beispielsweise als Bausteine der Biomembranen vorkommend, in Wasser, so bilden sich spontan geschlossene Membranstrukturen, sogenannte Vesikel. Da hier Temperatur und Druck gegeben sind (Wärmebad und Druckensemble) ist das thermodynamische Potential, das ein Minimum anstrebt die freie Enthalpie ΔG = ΔH − TΔS. Die Enthalpie ΔH lässt sich experimentell nachweisen, ist also messbar und ist positiv. Da der Prozess spontan abläuft, muss aber ΔG negativ sein, d. h. die Entropie muss steigen. Dies ist auf den ersten Blick verwirrend, da die Entropie meistens dafür verantwortlich ist, dass sich Substanzen vermischen (Mischungsentropie). Die Entropiezunahme liegt in einer besonderen Eigenschaft des Wassers begründet. Es bildet zwischen den einzelnen Wassermolekülen Wasserstoffbrückenbindungen aus, die ständig fluktuieren und somit einen hohen Beitrag zur Entropie des Wassers leisten. Um die langen Fettsäureketten der Lipide entsteht bei Lösung in Wasser ein größerer Bereich, in dem keine Wasserstoffbrückenbindungen mehr gebildet werden können. In den Bereichen um die Fettsäureketten herum fehlt der Entropiebeitrag der Wasserstoffbrücken, so dass die Entropie insgesamt abnimmt. Diese Abnahme ist erheblich größer als die durch das bloße Vermischen des Wassers und des Lipids zu erwartende Zunahme. Wenn sich die Fettsäureketten zusammenlagern, können mehr Wasserstoffbrücken gebildet werden, und die Entropie steigt. Man könnte dies auch so formulieren, dass die Fähigkeit des Wassers, fluktuierende Wasserstoffbrücken zu bilden, die Lipide aus der Lösung treibt. Letztlich ist diese Eigenschaft auch mit für die schlechte Löslichkeit vieler unpolarer Substanzen verantwortlich, die die Bildung von Wasserstoffbrückenbindungen stören.
Lebende Organismen: Ein lebender Organismus kann in gewissem Sinne als eine thermodynamische Maschine betrachtet werden, die chemische Energie in Arbeit und Wärme umwandelt und gleichzeitig Entropie zu produzieren scheint. Es ist nach dem gegenwärtigen Stand der Forschung noch nicht geklärt, ob einem biologischen System Entropie zuordenbar ist, da es sich nicht im Zustand des thermodynamischen Gleichgewichts befindet.
- Andere Disziplinen
Neben ihrer Rolle als fundamentale Zustandsgröße der phänomenologischen und statistischen Thermodynamik wird die Entropie in anderen Gebieten, insbesondere in der Informationstheorie und in der Wirtschaftswissenschaft benutzt. Die Entropie besitzt in diesen Gebieten eine eigenständige Bedeutung. So ist es z. B. in der Astrophysik notwendig, bei der Beschreibung von Sterngeburten, weißen Zwergen, Neutronensternen, schwarzen Löchern (sie haben die höchste Entropie aller bekannten physikalischen Systeme), Kugelsternhaufen, Galaxien(haufen) und letztendlich dem ganzen Kosmos, auf den Begriff der Entropie zurückzugreifen.
Statistische Physik
Um 1880 konnte Ludwig Boltzmann mit der von ihm und James Maxwell begründeten statistischen Physik auf mikroskopischer Ebene die Entropie erklären. In der statistischen Mechanik wird das Verhalten makroskopischer thermodynamischer Systeme durch das mikroskopische Verhalten seiner Komponenten, also Elementarteilchen und daraus zusammengesetzter Systeme wie Atome und Moleküle, erklärt. Ein Mikrozustand ist klassisch gegeben durch Angabe aller Orte und Impulse der zum System zählenden Teilchen. Ein solcher Mikrozustand
 ist demnach ein Element eines 6N-dimensionalen Vektorraums, der in diesem Zusammenhang Phasenraum genannt wird. Die kanonischen Gleichungen der klassischen Mechanik beschreiben die zeitliche Evolution des Systems, die Phasentrajektorie.
ist demnach ein Element eines 6N-dimensionalen Vektorraums, der in diesem Zusammenhang Phasenraum genannt wird. Die kanonischen Gleichungen der klassischen Mechanik beschreiben die zeitliche Evolution des Systems, die Phasentrajektorie.Alle unter gegebenen makroskopischen Randbedingungen, wie z. B. Gesamtenergie E, Volumen V und Teilchenzahl N, erreichbaren Phasenpunkte bilden ein zusammenhängendes Phasenraumvolumen Ω. Die Entropie ist ein Maß für das unter bestimmten makroskopischen Randbedingungen zugängliche Phasenraumvolumen, also für die Zahl der zugänglichen Zustände. Je größer die Entropie ist, desto unbestimmter ist der mikroskopische Zustand, desto weniger Informationen sind über das System bekannt. Das grundlegende Postulat der statistischen Physik besagt, dass jeder der zugänglichen Mikrozustände eines vollständig abgeschlossenen Systems im Gleichgewicht mit gleicher Wahrscheinlichkeit auftritt und somit die Entropie maximal ist (siehe: Maximum-Entropie-Methode, Mikrokanonischer Zustand).
Die Entropie ist proportional zum Logarithmus des zugänglichen Phasenraumvolumens (bzw. quantenmechanisch: der Zahl der zugänglichen Zustände) und berechnet sich im SI-System aus
in der Einheit J/K. Um diese Rechnung konkret ausführen zu können, müssen also zunächst die makroskopischen Observablen des betrachteten Systems bekannt sein. Für das ideale Gas ergibt sich die Sackur-Tetrode-Gleichung. Die Konstante kB wird in Anerkennung der Leistungen Ludwig Boltzmanns bei der Entwicklung der statistischen Theorie als Boltzmann-Konstante bezeichnet, er selbst hat ihren Wert jedoch nicht bestimmt.
Die Entropie der Thermodynamik ist die Shannon-Entropie der Verteilung der Zustände, welche die Shannon-Entropie maximiert (multipliziert mit einer Konstanten). Damit ist die thermodynamische Entropie nur ein Spezialfall für Gleichgewichtszustände mit maximaler Unkenntnis bei gegebenen Randbedingungen.
Quantenmechanik
In der Quantenstatistik ist ein Mikrozustand gegeben durch einen Vektor
 im Hilbertraum
im Hilbertraum  des Systems (typischerweise 1023 Teilchen!). Den zugehörigen Makrozustand beschreibt man durch einen statistischen Operator, der auch als Dichteoperator bezeichnet wird.
des Systems (typischerweise 1023 Teilchen!). Den zugehörigen Makrozustand beschreibt man durch einen statistischen Operator, der auch als Dichteoperator bezeichnet wird.Dieser enthält alle Informationen über das System, die durch eine ideale Messung zugänglich sind (das ist viel weniger als bei dem reinen Zustand
, dem Mikrozustand). Der Makrozustand ist klassisch gegeben durch ein Ensemble von Mikrozuständen, die mit als „typische makroskopische Werte“ bestimmte Erhaltungsgrößen gemein haben, wie z. B. Energie, Volumen und Teilchenzahl. Die Verteilung der Mikrozustände im Phasenraum ist klassisch durch eine Verteilungsfunktion gegeben, an deren Stelle in der quantenmechanischen Beschreibung der Dichteoperator tritt:pi ist die Wahrscheinlichkeit, dass sich das betrachtete System im quantenmechanischen Zustand
 befindet. Dabei ist jetzt die Zahl der betrachteten Freiheitsgrade gewöhnlich viel kleiner als
befindet. Dabei ist jetzt die Zahl der betrachteten Freiheitsgrade gewöhnlich viel kleiner als 
Der Erwartungswert einer Observablen auf dem durch den Dichteoperator beschriebenen Zustandsgemisch ist gegeben durch eine Spurbildung:
Die Spur eines Operators ist folgendermaßen definiert:
 für beliebiges vollständiges Orthonormalsystem
für beliebiges vollständiges Orthonormalsystem  .
.Die Entropie ist über die Wahrscheinlichkeiten der einzelnen reinen quantenmechanischen Zustände
im Makrozustand gegeben durchwobei pi die Wahrscheinlichkeit ist, im i-ten Mikrozustand zu sein. kB ist die Boltzmann-Konstante. Die Wahrscheinlichkeiten pi können Werte zwischen 0 und 1 annehmen, somit ist
 und die Entropie
und die Entropie  positiv semidefinit.
positiv semidefinit.Als Beispiel nehmen wir ein Spinsystem mit 4 Elektronen. Spin und magnetische Moment sind bekanntlich antiparallel; das magnetische Moment μ eines nach unten zeigenden Spins besitzt im äußeren Magnetfeld B die Energie − μB. Die Grundzustandenergie E0 des Systems soll insgesamt − 2μB sein, was auf folgende vier Zustände führt:
Daraus folgt, dass die Spinentartung
 ist und
ist und  gilt.
gilt.Die obige allgemeine Formel, (*), ist bis auf einen konstanten Faktor (der Ausdruck
 wird durch den sog. dualen Logarithmus ld ... ersetzt, den Logarithmus zur Basis 2) identisch mit der Formel für die Shannon'sche Informationsentropie. Das bedeutet, die physikalische Entropie ist auch ein Maß für die Information, die einem durch Kenntnis des Makrozustands zum Mikrozustand fehlt.
wird durch den sog. dualen Logarithmus ld ... ersetzt, den Logarithmus zur Basis 2) identisch mit der Formel für die Shannon'sche Informationsentropie. Das bedeutet, die physikalische Entropie ist auch ein Maß für die Information, die einem durch Kenntnis des Makrozustands zum Mikrozustand fehlt.Eigenschaften der statistischen Entropie eines quantenmechanischen Zustandes
Seien
 und
und  Dichteoperatoren auf dem Hilbertraum .
Dichteoperatoren auf dem Hilbertraum .- Invarianz unter unitären Transformationen von (mit
 )
)
- Minimum

- Minimum wird bei reinen Zuständen
 angenommen
angenommen
- Maximum

- Maximum wird angenommen, wenn alle möglichen Zustandsvektoren mit gleicher Wahrscheinlichkeit
 auftreten
auftreten
 mit
mit 
- Sei Dichteoperator auf
 und
und  bzw.
bzw.  reduzierte Dichteoperatoren auf
reduzierte Dichteoperatoren auf  bzw.
bzw. 

Entropie im Überblick
Die Thermodynamik beschreibt den Zustand einer Substanz mit Hilfe makroskopisch messbarer Größen wie Temperatur, Druck, innere Energie, Enthalpie, aber auch durch die relativen Mengen von Produkten und Edukten. Es gibt viele Zustandsgrößen und einige können als Funktionen anderer dargestellt werden (Zustandsfunktionen). Eine davon ist die Entropie (Einheit: Joule/Kelvin).
Während man aus der Alltagserfahrung mit Größen wie Länge, Energie oder Leistung und ihren Einheiten quantifizierbare Vorstellungen verbindet, ist die Einheit „Joule/Kelvin“ der Größe Entropie wenig anschaulich. Sie hat den Bedeutungsinhalt, dass „ein Zustand höherer Entropie sich auch mit höherer Wahrscheinlichkeit einstellt“. Der Begriff wurde ursprüngllich von Clausius geprägt und mit Hilfe thermodynamischer Größen quantifiziert, später dann auch von Boltzmann aus einer statistischen Überlegung heraus und mit Hilfe der Atom- bzw. Molekülvorstellung.
1. Statistische Formulierung
Was kann man sich nun unter der Wahrscheinlichkeit eines Zustands vorstellen? Eine Substanz besteht aus sehr vielen Teilchen. So wie bei einem Patienten derselbe Zustand (Temperatur, Pulsfrequenz) eine Vielzahl von Ursachen haben kann, die sich erst bei näherer Untersuchung herausstellen, so kann auch derselbe thermodynamische Zustand auf vielen und ganz unterschiedlichen Anordnungen der Moleküle beruhen. Würde man z.B. alle N Moleküle eines Gases nummerieren, dann könnte der Zustand der gleichmäßigen Verteilung über ein Volumen dadurch gebildet werden, dass sich alle Moleküle mit niedrigen Nummern (bis N/2) in der einen, die höheren Nummern in der anderen Hälfte aufhalten. Derselbe Zustand ergibt sich jedoch auch, wenn sich alle geraden bzw. ungeraden Nummern in der einen bzw. andern Hälfte befänden. Der Zustand der „gleichmäßigen Verteilung des Gases über das gesamte Volumen“ wird von einer außerordentlich großen Anzahl von Verteilungsmustern der Moleküle gebildet. Hingegen wird der Zustand, in dem sich die Moleküle auf ein Teilvolumen konzentrieren und der Rest leer bleibt, von deutlich weniger Anordnungen der Moleküle gebildet, wie im folgenden Beispiel:
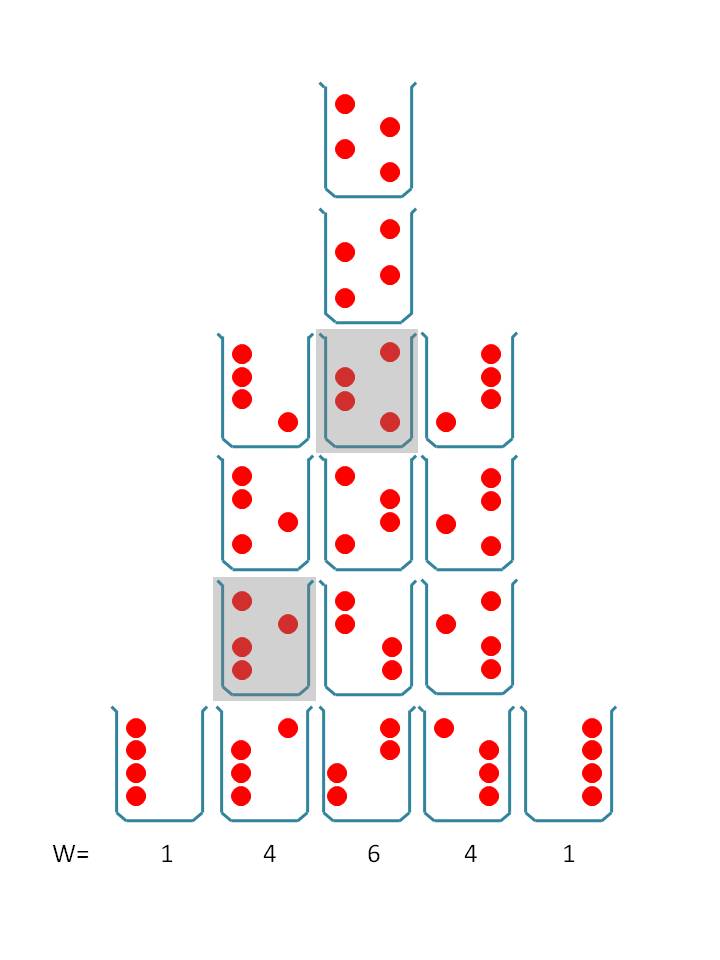
Entropie und die Verteilung im Raum
 Zu Säulen gestapelt sind alle Mikrozustände, die dem System dieselben makroskopischen Eigenschaften verleihen
Zu Säulen gestapelt sind alle Mikrozustände, die dem System dieselben makroskopischen Eigenschaften verleihenFrei bewegliche Moleküle verteilen sich gleichmäßig über einen Raum. Dieser Zustand weist die maximale Entropie auf, oder, anders ausgedrückt, er ist am wahrscheinlichsten. Das lässt sich an vier Molekülen zeigen (Abbildung), die in einem Becherglas ständig zwischen der linken und rechten Hälfte in Bewegung sind. (Um es auf das Wesentliche zu reduzieren, ist senkrechter Platztausch erst einmal nicht vorgesehen.) Es ergeben sich ganz unterschiedliche und stets wechselnde Verteilungen. In einer Momentaufnahme könnte z. B. nur das zweite Molekül von oben in der rechten Hälfte zu sehen sein, die restlichen drei links, in einer anderen die Moleküle zwei und drei auf der linken Seite (jeweils grau hervorgehoben). Jede einzelne Aufteilungsvariante – Mikrozustand genannt – ist gleich häufig oder wahrscheinlich. Zu Säulen gestapelt sind diejenigen Mikrozustände, bei denen sich auf einer Seite gleich viele Moleküle versammelt haben (keines bzw. maximal vier). Sie verleihen dem Gesamtsystem dieselben makroskopischen Eigenschaften (z. B. die Höhe des Drucks auf die linke oder rechte Wand) und bilden zusammen einen Makrozustand. Am häufigsten ist die gleichmäßige Aufteilung auf beide Hälften, denn sie kann durch die größte Anzahl von Mikrozuständen verwirklicht werden (in diesem Fall sechs). Sind mehr Moleküle im Becherglas, stehen mehr Raumzellen zur Verfügung oder lässt man auch senkrechten Platztausch zu, dann steigt die Vielfalt der Anordnungsmöglichkeiten rapide an. Aber immer hat eine gleichmäßige Aufteilung die höchste Anzahl von Mikrozuständen und stellt sich am häufigsten ein, auch wenn man anfangs die Moleküle, dicht gedrängt, nur in eine Ecke eingebracht hätte. Dies geschieht aus sich heraus und zufällig und nur deshalb, weil sich die Moleküle bewegen.
Die Anzahl der Mikrozustände eines Makrozustands heißt sein „Statistisches Gewicht W“. Das W kann, wenn es sich um viele Teilchen in der Gasphase handelt, eine sehr große Zahl sein (im Gegensatz zur mathematischen Wahrscheinlichkeit), kann aber auch sehr klein werden, wenn starke Kräfte zwischen den Teilchen wirken (Nullpunktsentropie). Der natürliche Logarithmus des statistischen Gewichts W eines Zustands, multipliziert mit der Boltzmann-Konstante kB, ergibt seine Entropie S:
Ändert man die Bedingungen, denen eine Substanz ausgesetzt ist (indem man z.B. das Volumen vergrößert), dann gehen die Moleküle in einen neuen Zustand über, nämlich in den, der unter den geänderten Bedingungen der wahrscheinlichste ist. Die Ursache dafür liegt in der Bewegung der Moleküle, die sich ständig neu über den Raum und die Energieniveaus verteilen. Die Annäherung an den Gleichgewichtszustand ist gekennzeichnet – nicht bewirkt – durch die Zunahme der Entropie. Diese ist das Maß für die Wahrscheinlichkeit eines Zustands. Die Entropie ist weder eine Energie noch eine Art von Substanz, sondern die Messgröße für einen Zustand höherer oder geringerer Wahrscheinlichkeit.
Über die spontane Einstellung des wahrscheinlichsten Makrozustands im Hinblick auf die räumliche Verteilung können der Joule-Thomson-Effekt, die Osmose und die Temperaturkonstanz beim Schmelzvorgang gedeutet werden.
Kurz: Allgemein ist ein Mikrozustand ein bestimmtes Verteilungsmuster der Moleküle, aber nicht nur im Raum, sondern auch über die Freiheitsgrade der Bewegung (z.B. Translation, Schwingungen und über deren Energieniveaus) und auch auf die Edukt- bzw. Produktmoleküle einer Reaktion. Viele Mikrozustände können denselben makroskopischen Zustand bilden, eben den „Makrozustand“, der dann durch makroskopisch messbare thermodynamische Größen charakterisiert werden kann. Da die Moleküle stets durcheinander schwirren, vergleichbar einem Bienenschwarm, sind alle Mikrozustände gleich häufig oder wahrscheinlich. Nicht aber die einzelnen Makrozustände, da sie von unterschiedlich vielen Mikrozuständen gebildet werden können. Das System nähert sich von selbst dem wahrscheinlichsten Zustand, dem mit der größten Anzahl von Mikrozuständen. Aus dem statistischen Gewicht W kann die Entropie berechnet werden. Sie ist ein (logarithmisches) Maß für die Anzahl der Arten W, auf die ein mikroskopisches System – im makroskopischen Sinn – gleich sein kann.
2. Thermodynamische Formulierung
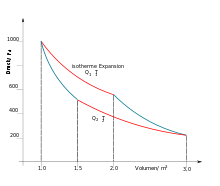 Carnot-Diagramm eines einatomigen Gases. Bei einer isothermen Expansion wird dem System die Wärmemenge Q1 bei der Temperatur T1 zugeführt, bei Kompression (untere rote Linie) gibt es Q2 bei T2 ab.
Carnot-Diagramm eines einatomigen Gases. Bei einer isothermen Expansion wird dem System die Wärmemenge Q1 bei der Temperatur T1 zugeführt, bei Kompression (untere rote Linie) gibt es Q2 bei T2 ab.Im Zusammenhang mit dem Carnot-Prozess findet man, dass umso mehr Wärme zwischen Reservoir und Arbeitsmedium fließt, je höher die Temperatur ist und umgekehrt. (Das ist mit Hilfe der Molekülvorstellung verständlich, da die Wärmeübertragung auf der Bewegungsenergie der Moleküle beruht und diese sich proportional zur absoluten Temperatur verhält.) Auch ohne die Molekülvorstellung fand Clausius diese Proportionalität, nämlich, dass im Carnot-Prozess die Größe Q/T, die reduzierte Wärme, beim Wärmezufluss (bei hoher Temperatur) und bei der Kühlung (bei tieferer Temperatur) des Arbeitsmediums den gleichen absoluten Wert hat und, zumindest im reversiblen Fall, in der Summe null ergibt.
Im ersten Schritt des Carnot-Prozesses, der isothermen Expansion, wird die zugeführte Wärmeenergie QT1 während der Ausdehnung des Arbeitsmediums in potenzielle Energie umgewandelt und so gespeichert (eine Anordnung, die dies leistet, beschreibt [2]). Unter Einsatz dieser gespeicherten Energie könnte der Vorgang wieder rückgängig gemacht werden. Gelingt diese Umwandlung von QT1 in Epot nicht vollständig, etwa weil Reibung Verluste bewirkt (irreversibler Fall), dann wird weniger Arbeit gespeichert (die nicht mehr für eine Umkehrung ausreichen würde) und die Differenz erscheint als zusätzliche (Reibungs-)Wärme, die abgegeben wird. Dieser Wärmebetrag zählt negativ und so ist die Summe aller Q/T-Beträge ebenfalls negativ. Je größer das Ausmaß der Irreversibilität ist, desto negativer ist die Summe. Damit gibt die Funktion Q/T auch eine quantitative Beschreibung der Irreversibilität. Clausius hat in diesem Zusammenhang die Funktion
 definiert, mit
definiert, mit  bzw.
bzw.  (irreversibel).
(irreversibel).
Kurz: Aus makroskopisch messbaren thermodynamischen Größen lässt sich eine Zustandsfunktion ableiten, die das Ausmaß der Irreversibiltiät wiedergibt.
3. Äquivalenz der Entropiebegriffe
Bei der Expansion eines Gases können zwei unterschiedliche Vorgänge vom gleichen Ausgangs- zum gleichen Endzustand führen:
a) Die isotherme Expansion im ständigen Gleichgewicht, unter Wärmezufuhr und äquivalenter Arbeitsleistung (erster Schritt im Carnot-Prozess). Von der zugeführten Wärmeenergie QT1 bleibt dabei nichts im Medium zurück; der Vorgang ist mit Hilfe der extern gespeicherten Energie umkehrbar.
b) Eine spontane isotherme Ausdehnung in einen größeren Raum (ins Vakuum), ohne Wärmezufuhr und ohne Arbeitsleistung (Versuch von Gay-Lussac). Es wird keine Wärme zugeführt, es wird keine Arbeit geleistet und auch nicht gespeichert. Wollte man aber den Vorgang umkehren, also isotherm komprimieren, dann müsste zusätzliche Arbeit aufgewendet werden, die dann als Wärme abgegeben wird. Also bei der Ausdehnung kein Wärmeeintrag ins Arbeitsmedium, aber Wärmeproduktion bei der Umkehrung; außerdem bleibt eine Veränderung in der Umgebung zurück, da die notwendige Arbeit beschafft werden muss. Ein irreversibler Vorgang.
Trotz der beiden unterschiedlichen Vorgehensweisen ist die Veränderung, die am Ende der isothermen Expansion am Arbeitsmedium bleibt, in beiden Fällen dieselbe. Mikroskopisch betrachtet weist der Endzustand eine größere Zahl von Mikrozuständen auf und hat damit ein größeres statistisches Gewicht W.
Der Wahrscheinlichkeit, ausgedrückt durch das statistische Gewicht, kommt in der statistischen Thermodynamik dieselbe Rolle zu wie der Entropie in der chemischen Thermodynamik. Es muss also eine Beziehung geben zwischen der Entropie und dem statistischen Gewicht. Um diesen zu finden, betrachtet man zwei unabhängige Systeme von Teilchen als ein Gesamtsystem. Dessen Entropie S muss gleich der Summe der einzelnen Entropien S1+S2 sein (Zustandsfunktionen). Die statistischen Gewichte W1 und W2 müssen jedoch multipliziert werden, da jeder Mikrozustand des einen mit jedem Mikrozustand des anderen Systems einen neuen Mikrozustand des Gesamtsystems bildet (W=W1∙W2). Diese beiden Forderungen sind erfüllt, wenn die Entropie eine logarithmische Funktion des statistischen Gewichts W ist:
Die Konstante k* ist erst einmal willkürlich gewählt. Es stellt sich heraus, dass sie gleich der Boltzmann-Konstanten kB = R/NA = 1,38∙10-23 Joule/Kelvin ist, wenn
a) W im thermodynamisch stabilen Zustand einen Maximalwert annimmt und b) die für den stabilen Zustand gültige Verteilungsfunktion die Boltzmann-Verteilung ist [3].
Damit hat S=k∙lnW dieselbe Dimension wie die Zustandsgröße Qrev/T, also Joule/Kelvin. Während mit makroskopisch messbaren Größen nur Entropiedifferenzen bestimmt werden können (z.B. vor und nach einer Erwärmung oder Reaktion), erlaubt die statistische Thermodynamik die Berechnung absoluter Entropien.
Kurz: Die mikroskopische und die makroskopische Vorgehensweise in der Thermodynamik führen auf denselben Entropiebegriff.
4. Berechnung und Verwendung tabellierter Entropiewerte
Die molare Entropie Smol bei einer bestimmten Temperatur T2 und bei konstantem Druck p erhält man mit Hilfe der molaren Wärmekapazität cp(T) durch Integration vom absoluten Nullpunkt bis zur aktuellen Temperatur:
Dazu kommen noch Entropieanteile bei Phasenübergängen. Nach Planck wird die Entropie ideal kristallisierter, reiner Festkörper am absoluten Nullpunkt gleich null gesetzt (Gemische oder frustierte Kristalle behalten dagegen eine Restentropie). Unter Standardbedingungen spricht man von der Standardentropie S0. Auch nach der statistischen Betrachtungsweise hängen Entropiewert und Wärmekapazität mit einander zusammen: Eine hohe Wärmekapazität bedeutet, dass ein Molekül viel Energie speichern kann, und das kann z.B. auf einer großen Zahl niedrig liegender und daher leicht erreichbarer Energieniveaus beruhen. Entsprechend viele unterschiedliche Verteilungsmöglichkeiten auf diese Niveaus gibt es dann auch für die Moleküle und das führt auch auf einen hohen Entropiewert für den wahrscheinlichsten Zustand.
In elektrochemischen Reaktionen ergibt sich die Reaktionsentropie ∆S aus der gemessene Änderung von dE (elektromotorische Kraft) mit der Temperatur:
 (z = Ladungszahl, F = Faraday-Konstante)
(z = Ladungszahl, F = Faraday-Konstante)
Die Entropieänderung bei idealen Mischungen erhält man mit Hilfe der Molenbrüche xi der beteiligten Substanzen:
wobei sich in realen Mischungen noch eine Zusatzentropie durch die Veränderung der zwischenmolekularen Kräfte beim Mischen ergibt.
Entstehen bei einer chemischen Reaktion neue Moleküle, dann tritt die höchste Entropie in einem ganz bestimmten Gleichgewichtszustand auf, bei dem sich die Moleküle sowohl auf die Edukt- wie auch auf die Produktniveaus verteilen können. Über die folgende Beziehung, in der die Differenzen der Standard-Entropiewerte ∆S0 der beteiligten Substanzen eine wesentliche Rolle spielen, kann die Gleichgewichtskonstante K berechnet werden:(das ∆ bedeutet in diesem Fall die Änderung der Größe bei vollständigem Reaktionsablauf). Woran man bei einem spontanen Vorgang (z.B. chemischen Reaktionen, Lösungs- und Mischungsvorgängen, Einstellung von Phasengleichgewichten und deren Temperaturabhängigkeit, Osmose u.a.) die Intensität dieses Vorgangs abschätzen kann, das ist die Zunahme der gesamten Entropie zwischen Anfangs- und Gleichgewichtszustand, die der Reaktanden und die der Umgebung zusammen genommen (->chemisches Gleichgewicht). Die spontane Zunahme der Entropie wiederum ist eine Folge der ständigen Bewegung der Moleküle.
Kurz: Die Standard-Entropie von Substanzen kann aus dem Verlauf der Wärmekapazität mit der Temperatur berechnet werden. Die Kenntnis tabellierter Entropiewerte ermöglicht (zusammen mit den Reaktonsenthalpien) die Voraussage des chemischen Gleichgewichts.
Literatur
- E-Books
- http://www.job-stiftung.de/index.php?id=8,22,0,0,1,0
- Georg Job, Regina Rüffler: Physikalische Chemie. Teil 1: Grundzüge der Stoffdynamik, Kapitel 2: Entropie und Temperatur (PDF-Datei; 3,16 MB)
- http://www.physikdidaktik.uni-karlsruhe.de/skripten/thermod.pdf (PDF-Datei; 12,87 MB)
- Grundlagen der Thermodynamik aufbauend auf der Entropie.
- http://www.egonsthermosite.eu/german_ttd/ger_ttd_scr/files/TTD_Kap_04_200.pdf, oder: http://web.me.com/egon.hassel/german_ttd/ger_ttd_scr/files/TTD_Kap_04_200.pdf: Entropiekapitel
- Lehrbücher und Übersichtsartikel
- Überblick über die verschiedenen Entropiebegriffe und deren Verknüpfungen: Frigg, R. and Werndl, C. "Entropy – A Guide for the Perplexed". In Probabilities in Physics; Beisbart C. and Hartmann, S. Eds; Oxford University Press, Oxford, 2010. PDF-Datei
- G. Adam, O. Hittmair: Wärmetheorie. 4. Auflage. Vieweg, 1992, ISBN 3-528-33311-1.
- Richard Becker: Theorie der Wärme. 3., erg. Auflage. Springer, 1985, ISBN 3-540-15383-7.
- Arieh Ben-Naim: Statistical Thermodynamics Based on Information: A Farewell to Entropy. 2008, ISBN 978-981-270-707-9
- Johan Diedrich Fast: Entropie. Huethig, 1982, ISBN 3-87145-299-8.
- Ulrich Nickel, Lehrbuch der Thermodynamik. Eine verständliche Einführung. 2. Auflage. PhysChem, 2011, ISBN 978-3-937744-06-3
- E. P. Hassel, T. V. Vasiltsova, T. Strenziok: Einführung in die Technische Thermodynamik; FVTR GmbH; Rostock 2010;ISBN 978-3-941554-02-3
- Arnold Sommerfeld: Vorlesungen über theoretische Physik – Thermodynamik und Statistik. Nachdruck der 2. Auflage. Harri Deutsch, 1988, ISBN 3-87144-378-6.
- Andre Thess: Das Entropieprinzip – Thermodynamik für Unzufriedene Oldenbourg-Wissenschaftsverlag, 2007, ISBN 3-486-58428-6.
- Handbuch der experimentellen Chemie Sekundarstufe II, Band 7: Chemische Energetik, Aulis Verlag Deubner GmbH & Co. KG, ISBN 978-3-7614-2385-1
- Populärwissenschaftliche Darstellungen
- Arieh Ben-Naim: Entropy Demystified - The Second Law Reduced to Plain Common Sense. New Jersey: World Scientific, Expanded Ed. 2008, ISBN 978-981-283-225-2 (populärwissenschaftliche, aber exakte Erklärung auf Grundlage der statistischen Physik).
- H. Dieter Zeh: Entropie. Fischer, 2005, ISBN 3-596-16127-4.
- Jeremy Rifkin, Ted Howard: Entropy: A New World View. Viking Press, New York 1980 (dt.: Entropie: Ein neues Weltbild. Hamburg, Hofmann & Campe, 1984).
Einzelnachweise und Anmerkungen
- ↑ Die verschiedenen Begriffe der Entropie werden etwa in Werndl und Frigg (2010) diskutiert.
- ↑ Wedler: Lehrbuch der Physikalischen Chemie, Verlag Chemie 1982. Abschnitt 1.1.16, S.46
- ↑ Wedler: Lehrbuch der Physikalischen Chemie, Verlag Chemie 1982. Abschnitt 4.2.1, S. 632
Weblinks
 Commons: Entropie – Sammlung von Bildern, Videos und Audiodateien
Commons: Entropie – Sammlung von Bildern, Videos und Audiodateien Wikibooks: Entropie – Lern- und Lehrmaterialien
Wikibooks: Entropie – Lern- und Lehrmaterialien Wikiquote: Entropie – Zitate
Wikiquote: Entropie – Zitate- Was ist Entropie? aus der Fernseh-Sendereihe alpha-Centauri
- Tomasz Downarowicz: „Entropy“. In: Scholarpedia (englisch, inkl. Literaturangaben)
- Martin Buchholz: Entropie - Von Kühltürmen und der Unumkehrbarkeit der Dinge Siegerbeitrag beim Science Slam Deutschland 2010 auf Youtube (Teil 1 von 2)
- Ulf von Rauchhaupt: Zeit, Tod und schmutziges Geschirr Artikel in der Frankfurter Allgemeinen Zeitung über Ludwig Boltzmann und die Entropie
- Was ist Entropie? (Publikationsmanuskript, A. Thess) (PDF-Datei; 107 kB)
- Thomas Neusius: Entropie und Richtung der Zeit Kursmaterial, beginnend auf Schulniveau
- Owen Maroney: Information Processing and Thermodynamic Entropy, in: Stanford Encyclopedia of Philosophy (englisch, inklusive Literaturangaben)
- Nico G. van Kampen: Entropie kurze, gut verständliche Erläuterung
- Welf A. Kreiner: Entropie – was ist das? Überblick für Studierende und Lehrer
Siehe auch
Kategorien:- Thermodynamische Zustandsgröße
- Physikalische Größenart




















![[\uparrow \downarrow \downarrow \downarrow]\,, \quad
[\downarrow \uparrow \downarrow \downarrow]\,, \quad
[\downarrow \downarrow \uparrow \downarrow]\,, \quad
[\downarrow \downarrow \downarrow \uparrow]\,.](b/a4b57e82a0bc63c51f167a1490093af5.png)










Wikimedia Foundation.