- Franz Hofmann (Pharmakologe)
-
Franz Bernhard Hofmann (* 21. Mai 1942 in Wien) ist ein deutscher Arzt und Pharmakologe.
 Franz Hofmann
Franz Hofmann
Inhaltsverzeichnis
Leben
Sein Vater war der Direktor des Instituts für anorganische und analytische Chemie der Technischen Hochschule Wien und später Direktor des anorganisch-chemischen Instituts der Ruprecht-Karls-Universität Heidelberg Ulrich Hofmann (1903–1986),[1] seine Mutter die Ärztin Renate Hofmann geb. Schiebeler. Schon der Großvater väterlicherseits war Chemiker, unter den früheren Vorfahren waren Juristen und Theologen. Franz besuchte bis zum Abitur 1962 das humanistische Ludwig-Georgs-Gymnasium in Darmstadt. Er studierte dann in Heidelberg, an der Ludwig-Maximilians-Universität München und an der Freien Universität Berlin Medizin. Seine Dissertation „Über die Wirkung einiger Colchizinderivate auf den Mäuse-Ascites-Tumor“ fertigte er bei Hans Lettré (1908–1971) an, dem Direktor des Heidelberger Instituts für Experimentelle Krebsforschung; 1968 wurde er zum Dr.med. promoviert.
Von 1970 bis 1985 arbeitete er an dem von Franz Gross (1913–1984) und anschließend Ulrich Schwabe (* 1935) geleiteten Heidelberger Pharmakologischen Institut. Unterbrochen wurde die Heidelberger Zeit von 1973 bis 1975 durch einen Aufenthalt am Department of Biological Chemistry der University of California, Davis bei Edwin Gerhard Krebs, der 1992 gemeinsam mit Edmond Henri Fischer für die Entdeckung der biologischen Steuerung durch reversible Proteinphosphorylierung den Nobelpreis für Physiologie oder Medizin erhielt. 1977 habilitierte sich Hofmann mit einer Arbeit „Charakterisierung der cAMP-abhängigen Protein-kinasen“ für Pharmakologie und Toxikologie. 1985 folgte er als Nachfolger von Volker Ullrich (* 1939) einem Ruf auf den Lehrstuhl für Physiologische Chemie der Universität des Saarlandes in Homburg (Saar) und 1990 als Nachfolger von Melchior Reiter (1919–2007) einem Ruf auf den Lehrstuhl für Pharmakologie und Toxikologie der Technischen Universität München, den er bis zu seiner Emeritierung 2008 innehatte. Während der Münchener Jahre war er von 1992 bis 1993 auch Gründungsdirektor des Berliner Forschungsinstituts für Molekulare Pharmakologie, das damals aus dem Institut für Wirkstoffforschung der Deutschen Demokratischen Republik hervorging. Von 1995 bis 2004 leitete er das Institut für Physiologische Chemie der Technischen Universität München kommissarisch.
Er ist verheiratet mit der Dermatologin Heidelore Hofmann geb. Schultze, mit der er einen Sohn hat.
Forschung
In den späten 1950er Jahren hatte Earl Wilbur Sutherland mit seiner Gruppe das cyclische Adenosinmonophosphat (cAMP) als einen second messenger bei der Wirkung chemischer Botenstoffe wie des Adrenalins entdeckt. „Diese Entdeckung legte mit einem Schlag die Barrieren zwischen der Pharmakologie und der Biochemie nieder, zum großen Vorteil beider Disziplinen.“[2] Seitdem sind die chemischen oder physikalisch-chemischen Reaktionskaskaden zwischen dem Kontakt von Pharmaka mit einer Zelle und der Antwort der Zelle, etwa einer Muskelkontraktion oder -erschlaffung, ein Hauptgegenstand der pharmakologischen Grundlagenforschung. Sie sind auch Franz Hofmanns Arbeitsgebiet, innerhalb dessen er sich auf Proteinkinasen, die durch cyclische Nukleotide reguliert werden, auf spannungsabhängige Calciumkanäle und auf Ionenkanäle, die durch cyclische Nukleotide reguliert werden, konzentriert.
cGMP-abhängige Proteinkinasen
Enzymproteine und Gene
Kurz bevor Hofmann 1970 im Heidelberger Pharmakologischen Institut begann, war (1963) ein zweites cyclisches Nukleotid entdeckt worden, das cyclische Guanosinmonophosphat (cGMP); hatte (1969) die Gruppe um Günter Schultz (* 1936) in Heidelberg ein cGMP-bildendes Enzym gefunden, eine Guanylylcylase, der cAMP-bildenden Adenylylcyclase analog;[3] und hatte (1968) Edwin Gerhard Krebs mit seiner Gruppe erkannt, dass cAMP eine cAMP-abhängige Proteinkinase, die Proteinkinase A, aktivierte und so die Phosphorylierung bestimmter Proteine vermehrte. Tat cGMP das gleiche? In den Worten Hofmanns und seines Doktoranden Guido Sold 1972: „... so far no system is known which is regulated by cGMP in mammalian tissues. In analogy to what has been found for cAMP one part of such a system could be a protein kinase regulated by cGMP. To examine such a possibility we studied the ability of cGMP and cAMP to stimulate the phosphorylation of histone by a protein kinase preparation from rat cerebellum.“ Die Antwort lautete: Ja, im Kleinhirn von Ratten gab es eine durch cGMP stimulierte, von der cAMP-stimulierten verschiedene Proteinkinase, und zwar im Cytoplasma gelöst.[4] Es war der erste Nachweis des Enzyms bei Säugern, nachdem es kurz zuvor bei Krebsen gefunden worden war. Hofmann erschloss sich damit ein Thema, das ihn bis heute (2011) [5] beschäftigt.
cGMP-abhängige Proteinkinasen wurden auch in anderen Organen gefunden, so im Herzen, in Thrombocyten und in glatter Muskulatur. Außer der gelösten Kinase, cGKI, gab es eine membrangebundene, cGKII, und von der löslichen gab es, bereits 1972 bemerkt, zwei durch alternatives Splicing gebildete Isoformen, cGKIα und cGKIβ, jede aus zwei identischen Untereinheiten bestehend. 1989 schließlich gelang es Hofmanns Gruppe gleichzeitig mit einer zweiten Gruppe, die cDNAs der beiden cGKI-Isoformen zu klonieren und damit ihre Aminosäuresequenz zu bestimmen.[6] Wenig später wurde in anderen Laboratorien die cGKII kloniert.
Funktionen
Die Frage nach den Funktionen der cGMP-abhängigen Proteinkinasen drängte besonders deshalb auf Antwort, weil 1977 festgestellt worden war, dass Nitrovasodilatatoren wie das Nitroglycerin, lange bekannte Arzneistoffe, die Blutgefäße durch Aktivierung der Guanylylcyclase und damit auf dem Weg über cGMP erweiterten. Darüber hinaus wurde 1980 ein den Nitrovasodilatatoren entsprechender körpereigener Botenstoff entdeckt, das Stickstoffmonoxid (NO). cGMP war also ein dem cAMP ebenbürtiger second messenger. Wirkte es in den Zellen – so wie cAMP über die Proteinkinase A – über die cGMP-abhängigen Proteinkinasen? Ließ es zum Beispiel so die glatte Muskulatur der Blutgefäße erschlaffen?
Hofmann griff zu der mit der Kenntnis der Gene gegebenen – molekulargenetischen – Möglichkeit, die Proteinkinasen durch Transfektion in Zellen einzuführen oder durch Gen-Knockout aus Zellen zu entfernen. Glatte Muskelzellen enthalten beide Isoformen der cGKI. Das Knockoutexperiment war eindeutig: Ausschaltung der cGKI bei Mäusen beseitigte die normale Relaxationswirkung von Stickstoffmonoxid und einem cGMP-ähnlichen Stoff auf die Blutgefäße, ohne die Relaxationswirkung eines cAMP-ähnlichen Stoffes zu beeinträchtigen. Außerdem war der arterielle Blutdruck der cGKI-defizienten Mäuse erhöht und reagierte nicht auf einen Nitrovasodilatator.[7]
Also war wirklich bei der Erweiterung von Blutgefäßen dem cGMP eine Aktivierung der cGKI nachgeschaltet. Die weiteren Schritte sind bis heute nicht ganz klar. Das entscheidende Signal für die Kontraktion glatter Muskelzellen ist ein Anstieg ihrer cytoplamatischen Calciumkonzentration. Calcium wird dabei unter anderem durch den second messenger Inositoltrisphosphat (IP3) aus dem intrazellulären endoplasmatischen Retikulum freigesetzt. Hofmanns Gruppe stellte fest, dass cGMP die IP3-vermittelte Freisetzung von Calcium via Aktivierung der cGKI hemmte. Als wichtigstes Ziel der Phosphorylierung bei dieser Hemmung wurde ein neues Protein identifiziert, das mit der cGKI und dem IP3-Rezeptor des endoplasmatischen Retikulums einen Komplex bildete und das die Autoren IRAG nannten, für IP3 receptor-associated cGMP kinase substrate.[8] Analog Deletion der cGKI verhinderte oder verminderte auch Deletion von IRAG bei Mäusen die normale Relaxationswirkung von Stickstoffmonoxid und einem cGMP-ähnlichen Stoff auf die Blutgefäße.[9]
Eine wichtige – aber keineswegs die einzige – Reaktionskaskade beim Kontakt von Stickstoffmonoxid oder Nitrovasodilatatoren mit Blutgefäßen lautete also:
„Stickstoffmonoxid → Aktivierung einer Guanylylcyclase → Bildung von cGMP → Aktivierung von cGKI → Phosphorylierung von IRAG → Hemmung der IP3-vermittelten Freisetzung von Calcium aus dem endoplasmatischen Retikulum → Gefäßerweiterung.“
Zusammengefasst haben sich folgende Funktionen der des cGMP → cGKI-Weges ergeben:[10]
- Erschlaffung der glatten Blutgefäßmuskulatur durch Stickstoffmonoxid und verwandte Stoffe, wie oben beschrieben, aber auch durch atriales natriuretisches Peptid (ANP) und verwandte Peptide, die ebenfalls eine Guanylylcyclase aktivieren;
- Erschlaffung der glatten Darmmuskulatur und damit Regelung der Darmtätigkeit; auch hier ist die IRAG-Kaskade beteiligt;
- Erschlaffung der glatten Muskulatur der Schwellkörper des Penis und damit Erektion; cGKI-defizient männliche Mäuse erzeugten viel weniger Nachkommen als normale;[11]
- Hemmung der Thrombocytenaggregation, eine weitere auch therapeutisch wichtige Wirkung von Stickstoffmonoxid, wieder über IRAG;[12]
- Hemmung der Freisetzung von Glucagon aus den Langerhans-Inseln des Pankreas;[5]
- Leitung des Wachstumskegels der Axone beim Wachstum von Nervenzellen;
- Sensibilisierung gegenüber Schmerzreizen;
- Anpassungen im Nervensystem wie die Langzeit-Depression im Kleinhirn.
Auch die cGKII hat Hofmanns Gruppe durch Gen-Knockout ausgeschaltet und folgende Funktionen des cGMP → cGKII-Weges gefunden:[10]
- Stimulation der Sekretion von Chloridionen und Wasser ins Darmlumen;
- Förderung der Knochenbildung; die cGKII-defizienten Mäuse blieben zwergwüchsig;[13]
- Anpassung der „inneren Uhr“ im Nucleus suprachiasmaticus an die äußere Zeit.
cAMP-abhängige Proteinkinase
Sein Aufenthalt an der University of California, Davis brachte Hofmann an die Quelle der cAMP-abhängigen Proteinkinase oder Proteinkinase A. Mit Edwin Gerhard Krebs gewann er neue Kenntnisse über das Enzym. Zwei Isoenzyme wurden gereinigt. Beide bestanden aus einem inaktiven Komplex von regulatorischen und katalytischen Untereinheiten, der in Gegenwart von cAMP zerfiel und die aktiven katalytischen Untereinheiten freigab.[14]
Zurück in Heidelberg, charakterisierte Hofmann das Enzym weiter und wies in Zusammenarbeit mit Wolfgang Trautwein vom Physiologischen Institut der Universität des Saarlandes eine seiner wichtigsten Funktionen direkt nach, nämlich die Vermittlung der positiv inotropen Herzwirkung von Adrenalin und anderen Substanzen, die β-Adrenozeptoren aktivieren, also β-Adrenozeptor-Agonisten. An isolierten Herzmuskelzellen steigerten Kontakt mit Adrenalin, Injektion von cAMP und Injektion der freien katalytischen Untereinheit der Proteinkinase in gleicher Weise den Einstrom von Calcium und die Kontraktionskraft. Die Reaktionskaskade lautete demnach:
„Adrenalin → β-Adrenozeptor → Aktivierung der Adenylylcyclase → Bildung von cAMP → Aktivierung der cAMP-abhängigen Proteinkinase → Steigerung des Calciumeinstroms → Steigerung der Kontraktionskraft.“[15]
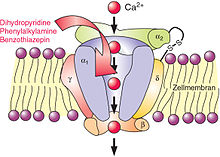 L-Typ-Calciumkanal mit (rot) drei Gruppen von Calciumantagonisten: Dihydropyridine (zu denen Nifedipin gehört), Phenylalkylamine (zu denen Verapamil gehört) und Benzothiazepine (zu denen Diltiazem gehört)[16]
L-Typ-Calciumkanal mit (rot) drei Gruppen von Calciumantagonisten: Dihydropyridine (zu denen Nifedipin gehört), Phenylalkylamine (zu denen Verapamil gehört) und Benzothiazepine (zu denen Diltiazem gehört)[16]Calciumkanäle
In die meisten zellulären Reaktionskaskaden sind Bewegungen von Calciumionen eingeschaltet. Besonders wichtig sind dabei spannungsabhängige (und zwar durch Depolarisation, also eine Verminderung des Membranpotentials, geöffnete) Calciumkanäle in der Zellmembran. Es gibt mehrere Typen, darunter die L-Typ-Kanäle (von long-lasting, mit langen Öffnungsdauern). Dass deren Öffnung in Herzmuskelzellen durch Aktivierung der cAMP-abhängigen Proteinkinase gefördert wurde, hatten Hofmann, Trautwein und ihre Kollegen in Heidelberg und Homburg 1982 nachgewiesen.[15] Doch blieben die Details unbekannt: „The nature and localization of the proteins phosphorylated by <the cAMP-dependent protein kinase> have not been determined.“ Ähnlich drei Jahre später: „The final step of the cascade, i.e. phosphorylation of a Ca channel-related protein, however, remains still controversial with respect to the nature of the protein and its relation to the Ca channel.“[17]
In Heidelberg arbeitete Hofmann mit dem Physiologen Johann Caspar Rüegg (* 1930) zusammen, der sich für das Calcium in der glatten Muskulatur interessierte. In Homburg war Wolfgang Trautwein physiologischer Partner. Außerdem stand ihm der Pharmakologe Hartmut Glossmann (* 1940) in Gießen wissenschaftlich nah. Glossmann forschte über Calciumantagonisten wie Verapamil, Nifedipin und Diltiazem, die die L-Calciumkanäle blockierten und deshalb zum Beispiel bei koronarer Herzkrankheit verwendet wurden. Er hatte 1981 erstmals radioaktiv markierte Calciumantagonisten zur Untersuchung von L-Typ-Calciumkanälen eingesetzt.
Kanalproteine und Gene
Diese Umgebung förderte, was 1985 in Homburg begann, nämlich die Reinigung der Bindungsstellen für radioaktiv markierte Calciumantagonisten im Gewebe,[18] ihre Identifizierung mit dem größten von vier Proteinen der L-Typ-Calciumkanäle und die Rekombination der gereinigten Proteine in künstlichen Lipiddoppelschichten zu funktionierenden Kanälen.[19] Der Proteinchemie folgte wieder die molekulare Genetik. Nachdem japanische und US-amerikanische Laboratorien 1987 und 1988 die größte Untereinheit, α1, und eine zweite, α2δ, kloniert hatten, klonierte Hofmanns Gruppe 1989 die cDNA der β-Untereinheit[20] und 1990 die cDNA der γ-Untereinheit:[21] mit α1/α2δ/β/γ war der Kanal komplett.
Für die cDNA-Klonierung hatten alle Forschergruppen ihres besonders hohen Gehalts an L-Typ-Calciumkanälen wegen Skelettmuskeln von Kaninchen verwendet. Andere Geweben schlossen sich an. Sie enthielten verwandte, aber nicht unbedingt identische Untereinheiten, und insgesamt kennt man heute für die große Familie der spannungsabhängigen Calciumkanäle 11 α1-Gene (und damit α1-Proteine), 4 α2δ-Gene (und damit α2δ-Proteine), 4 β-Gene (und damit β-Proteine) und 4 γ-Gene (und damit γ-Proteine), eine molekulare Vielfalt, die die Vielfalt der physiologischen und pharmakologischen Eigenschaften der Kanäle widerspiegelt. Die α1-Untereinheit bildet die Pore und enthält die spannungsempfindlichen Aminosäuren sowie – bei L-Typ-Kanälen – die Bindungsstellen für Calciumantagonisten. Die zuerst klonierten Kanäle in der Skelettmuskulatur heißen in heutiger Nomenklatur Cav1.1, die wichtigsten Kanäle in Herz und glatter Muskulatur Cav1.2., einige Kanäle in Sinneszellen Cav1.3.[22][23]
Funktionen
Grundsätzlich ist die physiologische Bedeutung der Kanäle lange bekannt: Enthält der Extrazellularraum kein Calcium, hört das Herz auf zu schlagen. Molekulargenetische Arbeiten haben viele Einzelheiten ans Licht gebracht.
- Der Cav1.3-Calciumkanal vermittelt in der Hörschnecke die Freisetzung des Neurotransmitters der inneren Haarzellen und ist deshalb notwendig für das Hören. Eine Gruppe im Institut von Hartmut Glossmann in Innsbruck fand, dass Ausschaltung des Gens dieser α1-Untereinheit bei Mäusen zu Taubheit führte.[24]
- Der Cav1.2-Kanal ist essentiell für Herz und glatte Muskulatur. Mäuse mit vollständigem Gen-Knockout starben noch in utero mangels Herztätigkeit. Auch nach selektivem („konditionalem“) Knockout in der glatten Muskulatur waren die Tiere schwer krank und starben bald an Darmverschluss durch Lähmung der Darmmuskulatur. Außerdem war der Blutdruck war zu niedrig und reagierte nicht auf Vasokonstriktoren wie Noradrenalin.[25]
- Der Cav1.2-Kanal trägt auch bei zur Freisetzung von Insulin aus den β-Zellen der Bauchspeicheldrüse und
- zu Anpassungen im Zentralnervensystem wie der Langzeit-Potenzierung im Corpus amygdaloideum.[26]
Ein Problem jedoch, das ganz am Anfang stand, ist bis heute nicht gelöst: das 1982[15] und dann wieder 1985[17] formulierte Problem der Reaktionskaskade zwischen den β-Adrenozeptoren des Herzenmuskels und der Steigerung des Calciumeinstroms durch die L-Typ-Kanäle und damit der Kontraktionskraft. 2008 bestätigte die Münchener Gruppe zwar, dass die cAMP-abhängige Proteinkinase die α1-Untereinheit des Cav1.2-Kanals in Herzmuskelzellen von Mäusen an einem bestimmten Serin, dem Serin 1298, phosphorylierte. Verhinderung der Phosporylierung durch Austausch des Serins gegen die nicht-phosphorylierbare Aminosäure Alanin verminderte aber die Steigerung von Calciumeinstrom und Kontraktionskraft durch den β-Adrenozeptor-Agonisten Isoprenalin nicht. Zur β-Adrenozeptor-Kaskade gehörte zweifellos eine Phosphorylierung durch die cAMP-abhängige Kinase, aber: „What is the physiological substrate of <the cAMP kinase> in the Cav1.2 channel complex?“[27] Die Frage besteht fort.
 Synthese und Rezeptoren von cGMP. NO aktiviert die lösliche (GCs), ANP eine membrangebundene Guanylylcyclase (GCp). cGMP wirkt auf Phosphodiesterasen (PDE), auf eine cGMP-Kinase und auf CNG-Kanäle. „Mittels dieser CNG-Kanäle sehen Sie diese Abbildung.“[28]
Synthese und Rezeptoren von cGMP. NO aktiviert die lösliche (GCs), ANP eine membrangebundene Guanylylcyclase (GCp). cGMP wirkt auf Phosphodiesterasen (PDE), auf eine cGMP-Kinase und auf CNG-Kanäle. „Mittels dieser CNG-Kanäle sehen Sie diese Abbildung.“[28]Durch cyclische Nukleotide gesteuerte Kanäle
Die cAMP- und cGMP-abhängigen Proteinkinasen sind nicht die einzigen Moleküle, die cAMP- und cGMP-Signalkaskaden fortsetzen. Es gibt zwei Gruppen von Kationenkanälen, deren Öffnung durch direkte Bindung von cAMP oder cGMP gefördert wird, ohne dass eine Phosphorylierung stattfände: die CNG-Kanäle, von cyclic nucleotide-gated, und die HCN-Kanäle, von hyperpolarization-activated, cyclic nucleotide-gated. Die Bindungsstelle ist eine intrazelluläre Aminosäuresequenz nah dem C-Terminus der Kanal-Untereinheiten, die CNBD, cyclic nucleotide-binding domain.[29][30]
CNG-Kanäle
Seit 1985 weiß man, dass cGMP in den Stäbchen und Zapfen der Netzhaut des Auges die Wahrnehmung von Licht vermittelt, und seit 1987 weiß man, dass cAMP in den Riechzellen der Nase die Wahrnehmung von Duftstoffen vermittelt. Die cyclischen Nukleotide binden sich in den Sinneszellen direkt an CNG-Kanäle und öffnen sie; gegenüber Änderungen des Membranpotentials sind die Kanäle, anders als zum Beispiel die spannungsabhängigen Calciumkanäle, kaum empfindlich.
Die erste Untereinheit eines CNG-Kanals, CNGA1, wurde 1989 von einer japanisch-deutschen Arbeitsgruppe aus der Netzhaut von Rindern kloniert, die zweite, CNGA2, 1990 von einer US-amerikanischen Arbeitsgruppe aus der Riechschleimhaut von Ratten. CNGA1 ist eine Untereinheit in den Netzhaut-Stäbchen, nicht den (für das Farbensehen zuständigen) Netzhaut-Zapfen. Der erste Beitrag von Hofmanns Gruppe kam 1994. Ausgehend von den bekannten Nukleotidsequenzen der CNGA1- und CNGA2-Gene, also a priori molekulargenetisch, klonierte sie die cDNA einer Untereinheit, CNGA3, in einem ganz anderen Gewebe, nämlich der Niere von Rindern.[31] Außer in der Niere kam die Untereinheit in Herz und Hoden vor, und bald stellte sich ihre Identität mit einer 1993 in den Zapfen der Netzhaut von Hühnern gefundenen Untereinheit heraus. Insgesamt kennt man heute sechs CNG-Untereinheiten, die sich für die Kanalbildung zu Tetrameren zusammenlegen.[30] Die Funktion der Kanäle in Nicht-Sinneszellen ist unklar. Vielleicht löst cGMP durch ihre Öffnung einen Einstrom von Calciumionen aus.
Ausschaltung des CNGA3-Gens bei Mäusen störte ihr Wachstum, ihr Verhalten und ihre Fortpflanzung kaum, hatte aber eine schwerwiegende Folge: Die Zapfen ihrer Netzhaut waren kurz nach der Geburt vermindert und verschwanden im Alter von acht Monaten ganz; jede Zapfenfunktion fehlte. Vermenschlichend ausgedrückt: die Tiere waren farbenblind. In der Tat beruht die Farbenblindheit mancher Menschen auf einer Mutation im CNGA3-Gen.[32]
In einem Lehrbuch-Schema der intrazellulären Wirkorte von cGMP hat Hofmann zu den CNG-Kanälen kommentiert: „Mittels dieser CNG-Kanäle sehen Sie diese Abbildung.“[28]
HCN-Kanäle
Durch cAMP wird auch die Öffnung einer Gruppe von Kationenkanälen gefördert, die ihren Erforschern so merkwürdig schien, dass sie die elektrischen Ströme durch die Kanäle If (funny) oder Iq (queer) nannten. Eine dritte Benennung Ih leitet sich von eben dieser Merkwürdigkeit ab: nämlich dass die Kanäle nicht wie die meisten spannungsabhängigen Ionenkanäle durch Depolarisation (Verminderung des Membranpotenials) geöffnet werden, sondern durch Hyperpolarisation (Erhöhung des Membranpotentials); die Kanalöffnung bewirkt dann Depolarisation durch Einstrom von Natriumionen. Seit dem Ende der 1970er Jahre vermutet man in den Strömen eine oder die Ursache des rhythmischen Feuerns von Aktionspotentialen in Nervenzellen und, besonders faszinierend, in den Sinusknotenzellen des Herzens, die den Herzschlag initiieren.
In Hofmanns Münchener Gruppe ging man davon aus, cAMP könnte sich in den If/q/h-Kanälen an eine ähnliche CNBD binden wie in den CNG-Kanälen. Beginnend mit einer computergestützten Suche in Nukleotidsequenz-Datenbanken entdeckte die Gruppe in der Tat 1998 und 1999 im Gehirn von Mäusen und im Herzen von Menschen vier neue Gene sowie die entsprechenden Proteine für die If/q/h-Kanäle.[33][34] Gleichzeitig gelang die Entdeckung einer US-amerikanischen und einer zweiten deutschen Gruppe. Die beiden Charakteristika Öffnung durch Hyperpolarisation und Öffnungsförderung durch cyclische Nukleotide führten zu der Bezeichnung HCN-Kanäle, HCN1 bis HCN4, hyperpolarization-activated, cyclic nucleotide-gated. In den Zellmembanen lagern sich die Untereinheiten wie bei den CNG-Kanälen für die Porenbildung zu Tetrameren, meist aus verschiedenen Untereinheiten bestehend, zusammen.[30] [35]
HCN-Kanäle gibt es im Wesentlichen in Nervenzellen und im Herzen. Im Gehirn ist besonders HCN2 weit verbreitet. Im Sinusknoten des Herzens von Menschen und Mäusen bilden HCN4 und HCN2 die Tetramere (80% HCN4 und 20% HCN2). Auch das nachgeschaltete Erregungsleitungssystem des Herzens besitzt HCN-Kanäle.
Im Gehirn tragen HCN-Kanäle zu Anpassungen wie Langzeit-Potenzierung, Lernen und Gedächtnis bei.[35] Mäusen mit einer Deletion des HCN2-Gens fehlte in den vom Thalamus zur Großhirnrinde ziehenden (thalamo-kortikalen) Neuronen der Ih-Strom. Es fehlte also ein depolarisierender Strom, und dadurch war das Ruhemembranpotential erhöht. Verminderte Bewegungsaktivität und das Elektroenzephalogramm zeigten eine Absence-Epilepsie.[36] Auch menschliche Absence-Epilepsie resultiert aus einer Hyperpolarisation thalamo-kortikaler Neurone[37] – eine Bedeutung von HCN2 für die Krankheit liegt nahe.
 Schrittmacher-Aktionspotential mit zugrundeliegenden Ionenströmen
Schrittmacher-Aktionspotential mit zugrundeliegenden IonenströmenWas schließlich die HCN-Kanäle im Herzen angeht, so geben sie der Forschung Rätsel auf. Die faszinierende Vermutung lautete, sie seien hauptverantwortlich für die rhythmischen Aktionspotentiale im Sinusknoten, die Schrittmacher-Aktionspotentiale, und dank der Förderung ihrer Öffnung durch cAMP für die positiv chronotrope Wirkung einer β-Adrenozeptor-Aktivierung – so wie die cAMP-Proteinkinase für deren positiv inotrope Wirkung hauptverantwortlich ist. Bei den HCN2-Knockout-Mäusen waren aber die mittlere Herzfrequenz und die Beschleunigung durch cAMP und Isoprenalin unverändert; der Herzschlag war nur weniger regelmäßig.[36]
Mäuse mit einer Deletion des HCN4-Gens, des Gens für die überwiegende HCN-Untereinheit der Sinusknotenzellen, starben in utero etwa am 10. Embryonaltag. Vor dem Tod entnommene Herzen schlugen verlangsamt, und die normalen, reifen Schrittmacher-Aktionspotentiale fehlten. Anscheinend genügte der mangels reifer Schrittmachertätigkeit zu langsame Herzschlag für die Blutversorgung des Embryos nach dem 10. Tag nicht.[38] Eine große Überraschung kam aber, „a big surprise“,[35] als HCN4 durch konditionalen Knockout erst nach der Geburt ausgeschaltet wurde, bei 8 Wochen alten Mäusen. Der Ih-Strom war dann stark vermindert, durch die Verminderung eines depolarisierenden Stromes waren die Sinusknotenzellen leicht hyperpolarisiert, und zuweilen stoppte das Herz (für einige Zehntelsekunden). Im übrigen aber lebten die Mäuse anscheinend unbeeinträchtigt, ihre mittlere Herzfrequenz war unverändert, Isoprenalin steigerte sie normal (auf maximal 700 pro Minute), und in Gegenwart von Isoprenalin bildeten die Sinusknotenzellen reife Schrittmacher-Aktionspotentiale.[39]
Was ersetzte die fehlende HCN4-Untereinheit bei der Bildung normaler Schrittmacher-Aktionspotentiale? Was, wenn nicht HCN4 oder HCN2, setzte β-Adrenozeptor-Aktivierung und cAMP in Herzbeschleunigung um? Das Rätsel wird zum Paradox, nimmt man hinzu, dass bei Menschen mit einer Mutation im HCN4-Gen die Herzfrequenz sehr wohl stark erniedrigt war und nicht auf cAMP reagierte. Woher der Speciesunterschied?[35] Wie die Frage nach den Schritten vom cAMP zur Steigerung der Kontraktionskraft des Herzens (siehe oben) besteht auch die Frage nach den Schritten vom cAMP zur Steigerung der Herzfrequenz fort.
Inzwischen haben die HCN-Kanäle des Herzens therapeutische Bedeutung erlangt. 1979 wurde ein Abkömmling des Clonidins beschrieben, St 567 oder Alinidin, der selektiv das Herz verlangsamte.[40]. Er wirkt, wie man heute weiß, durch Blockade der HCN-Kanäle im Sinusknoten. Ein anderer „Sinusknoten-Inhibitor“, Ivabradin, wird seit 2006 bei koronarer Herzkranheit angewandt.[35]
Lehre
Seit 2001 schreibt Hofmann das Kapitel „Wirkungen von Pharmaka auf den Organismus: allgemeine Pharmakodynamik“ des 2001 von Wolfgang Forth, Dietrich Henschler, Walter Rummel, Ulrich Förstermann und Klaus Starke herausgegebenen Lehrbuchs „Allgemeine und spezielle Pharmakologie und Toxikologie“. Seit 2005 ist er Mitherausgeber, zuletzt der 10. Auflage.[41]
Seit 2003 schreibt er die Kapitel „Smooth muscle tone regulation“ und „Voltage-dependent Ca2+ channels“ der „Encyclopedic Reference of Molecular Pharmacology“.[42]
Schüler
Folgende Wissenschaftler haben sich bei Franz Hofmann habilitiert (mit Jahr der Habilitation):
- Veit Flockerzi (1987), später Lehrstuhlinhaber für experimentelle und klinische Pharmakologie und Toxikologie in Homburg (Saar)
- Peter Ruth (1994), später Lehrstuhlinhaber für Pharmakologie und Toxikologie in Tübingen
- Martin Biel (1995), später Lehrstuhlinhaber für Pharmakologie für Naturwissenschaften der Ludwig-Maximilians-Universität München
- Wolfgang Dostmann (1996), später associate Professor of Pharmacology der University of Vermont in Burlington (Vermont)
- Alexander Pfeifer (1997), später Lehrstuhlinhaber für Pharmakologie und Toxikologie in Bonn
- Norbert Klugbauer (1998), später Professor für Pharmakologie in Freiburg im Breisgau
- Thomas Kleppisch (1999)
- Xiangang Zong (1999)
- Andreas Ludwig (2000), später Lehrstuhlinhaber für Pharmakologie und Toxikologie in Erlangen
- Gerhard Rammes (2001)
- Jens Schlossmann (2001), später Proferssor für Pharmakologie und Toxikologie in Regensburg
- Robert Feil (2003), später Professor am Interdisziplinären Institut für Biochemie Tübingen
- Andrea Welling (2003)
- Jörg Wegener (2004)
- Horst Thiermann (2005)
- Juliane Stieber (2006)
- Sven Moosmang (2006)
Anerkennung
Im Jahr 1998 ernannte die Tongji Medical University in Wuhan, 2004 die Chinesische Akademie der Wissenschaften Hofmann zum Professor honoris causa. Seit 2001 ist er Mitglied der Bayerischen Akademie der Wissenschaften, seit 2002 Mitglied der Deutschen Akademie der Naturforscher Leopoldina und seit 2003 Mitglied der Academia Europaea. 2002 hielt er in Freiburg im Breisgau die Ludwig Aschoff-Vorlesung der Freiburger Medizinischen Gesellschaft. Ebenfalls 2002 erhielt er den Max-Planck-Forschungspreis, 2003 den Preis der Feldberg Foundation, 2004 das Verdienstkreuz 1. Klasse der Bundesrepublik Deutschland und 2006 den Bayerischen Verdienstorden.
Einzelnachweise
- ↑ Hanns-Peter Boehm: Ulrich Hofmann 1903–1986. In: Chemische Berichte. 120, 1987, S. XXXVII–L. doi:10.1002/cber.19871201224. Abgerufen am 28. Mai 2011.
- ↑ H.P. Rang und M.M. Dale: Pharmacology, 2. Auflage. Edinburgh, Churchill Livingstone 1991, S. 38. ISBN 0-443-04110-5
- ↑ E. Böhme, K. Munske und G. Schultz: Bildung von cyclischem Guanosin-3‘,5‘-monophosphat in verschiedenen Geweben der Ratte. In: Naunyn-Schmiedebergs Archiv für experimentelle Pathologie und Pharmakologie. 264, 1969, S. 220–221
- ↑ Franz Hofmann und Guido Sold: A protein kinase activity from rat cerebellum stimulated by guanosine-3‘:5‘-monophosphate. In: Biochemical and Biophysical Research Communications. 49, 1972, S. 1100–1107. doi:10.1016/0006-291X(72)90326-9.
- ↑ a b Veronika Leiss, Andreas Friebe, Andrea Welling, Franz Hofmann und Robert Lukowski: Cyclic GMP kinase I modulates glucagon release from pancreatic α-cells. In: Diabetes. 60, 2011, S. 148–156. doi:10.2337/db10-0595. Abgerufen am 1. Juni 2011.
- ↑ W. Wernet, V. Flockerzi und F. Hofmann: The cDNA of the two isoforms of bovine cGMP-dependent protein kinase. In: FEBS Letters. 251, 1989, S. 191-196. doi:10.1016/0014-5793(89)81453-X. Abgerufen am 1. Juni 2011.
- ↑ Alexander Pfeifer, Peter Klatt, Steffen Massberg, Lars Ny, Matthias Sausbier, Christoph Hirneiß, Ge-Xing Wang, Michael Korth, Attila Aszódi, Karl-Erik Andersson, Fritz Krombach, Artur Mayerhofer, Peter Ruth, Reinhard Fässler und Franz Hofmann: Defective smooth muscle regulation in cGMP kinase I-deficient mice. In: EMBO Journal. 17, 1998, S. 3045–3051. doi:10.1093/emboj/17.11.3045. Abgerufen am 1. Juni 2011.
- ↑ Jens Schlossmann, Aldo Ammendola, Keith Ashman, Xiangang Zong, Andrea Huber, Gitte Neubauer, Ge-Xing Wang, Hans-Dieter Allescher, Michael Korth, Matthias Wilm, Franz Hofmann und Peter Ruth: Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Iβ. In: Nature. 404, 2000, S. 197–201. doi:10.1038/35004606. Abgerufen am 1. Juni 2011.
- ↑ Matthias Desch, Katja Sigl, Bernhard Hieke, Katharina Salb, Frieder Kees, Dominik Bernhard, Angela Jochim, Beate Spiessberger, Klaus Höcherl, Robert Feil, Susanne Feil, Robert Lukowski, Jörg W. Wegener, Franz Hofmann und Jens Schlossmann: IRAG determines nitric oxide- and atrial natriuretic peptide-mediated smooth muscle relaxation. In: Cardiovascular Research. 86, 2010, S. 496–505. doi:10.1093/cvr/cvq008. Abgerufen am 31. Mai 2011.
- ↑ a b F. Hofmann, R. Feil, T. Kleppisch und J. Schlossmann: Function of cGMP-dependent protein kinases as revealed by gene deletion. In: Physiological Reviews. 86, 2006, S. 1–23. doi:10.1152/physrev.00015.2005. Abgerufen am 31. Mai 2011.
- ↑ Petter Hedlund, Attila Aszódi, Alexander Pfeifer, Per Alm, Franz Hofmann, Marianne Ahmad, Reinhard Fässler und Karl-Erik Andersson: Erectile dysfunction in cyclic GMP-dependent kinase I-deficient mice. In: Proceedings of the National Academy of Sciences. 97, 2000, S. 2349–2354. doi:10.1073/pnas.030419997. Abgerufen am 31. Mai 2011.
- ↑ Melanie Antl, Marie-Luise von Brühl, Christina Eiglsperger, Matthias Werner, Ildiko Konrad, Thomas Kocher, Matthias Wilm, Franz Hofmann, Steffen Massberg und Jens Schlossmann: IRAG mediates NO/cGMP-dependent inhibition of platelet aggregation and thrombus formation. In: Blood. 109, 2007, S. 552–559. doi:10.1182/blood-2005-10-026294. Abgerufen am 31. Mai 2011.
- ↑ Alexander Pfeifer, Attila Aszódi, Ursula Seidler, Peter Ruth, Franz Hofmann und Reinhard Fässler: Intestinal secretory defects and dwarfism in mice lacking cGMP-dependent protein kinase II. In: Science. 274, 1996, S. 2082–2086. doi:10.1126/science.274.5295.2082. Abgerufen am 1. Juni 2011.
- ↑ Franz Hofmann, Joseph A. Beavo, Peter J. Bechtel und Edwin G. Krebs: Comparison of adenosine 3':5'-monophosphate-dependent protein kinase from rabbit skeletal muscle and bovine heart muscle. In: The Journal of Biological Chemistry. 250, 1975, S. 7795–7801.
- ↑ a b c W. Osterrieder, G. Brum, J. Hescheler, W. Trautwein, V. Flockerzi und F. Hofmann: Injection of subunits of cyclic AMP-dependent protein kinase into cardiac myocytes modulates Ca2+ current. In: Nature. 298, 1982, S. 576–578. doi:10.1038/298576a0. Abgerufen am 6. Juni 2011.
- ↑ U. Förstermann: Pharmakologie des cardiovaskulären Systems. In: K. Aktories, U. Förstermann, F. Hofmann und K. Starke: Allgemeine und spezielle Pharmakologie und Toxikologie. 10. Auflage, München, Elsevier GmbH 2009, Seite 449-485. ISBN 978-3-437-42522-6
- ↑ a b M. Kameyama, F. Hofmann und W. Trautwein: On the mechanism of β-adrenergic regulation of the Ca channel in the guinea-pig heart. In: Pflügers Archiv. 405, 1985, S. 285–293. doi:10.1007/BF00582573. Abgerufen am 12. Juni 2011.
- ↑ Peter Ruth, Veit Flockerzi, Egbert von Nettelbladt, Jochem Oeken und Franz Hofmann: Characterization of the binding sites for nimodipine and (-)-desmethylmethoxyverapamil in bovine cardiac sarcolemma. In: European Journal of Biochemistry. 150, 1985, S. 313–322. doi:10.1111/j.1432-1033.1985.tb09023.x. Abgerufen am 12. Juni 2011.
- ↑ Franz Hofmann, Wolfgang Nastainczyk, Axel Röhrkasten, Toni Schneider und Manfred Sieber: Regulation of the L-type calcium channel. In: Trends in Pharmacological Sciences. 8, 1987, S. 393–398. doi:10.1016/0165-6147(87)90103-9. Abgerufen am 12. Juni 2011.
- ↑ Peter Ruth, Axel Röhrkasten, Martin Biel, Eva Bosse, Stefan Regulla, Helmut E. Meyer, Veit Flockerzi und Franz Hofmann: Primary structure of the β subunit of the DHP-sensitive calcium channel from skeletal muscle. In: Science. 245, 1989, S. 1115–1118. doi:10.1126/science.2549640. Abgerufen am 12. Juni 2011.
- ↑ E. Bosse, S. Regulla, M. Biel, P. Ruth, H.E. Meyer, V. Flockerzi und Franz Hofmann: The cDNA and deduced amino acid sequence of the γ subunit of the L-type calcium channel from rabbit skeletal muscle. In: FEBS Letters. 267, 1990, S. 153–156. doi:10.1016/0014-5793(90)80312-7. Abgerufen am 13. Juni 2011.
- ↑ F. Hofmann, M. Biel und V. Flockerzi: Molecular basis for Ca2+ channel diversity. In: Annual Review of Neuroscience. 17, 1994, S. 399–418. doi:10.1146/annurev.ne.17.030194.002151. Abgerufen am 13. Juni 2011.
- ↑ William A. Catterall, Edward Perez-Reyes, Terrance P. Snutch und Joerg Striessnig: International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. In: Pharmacological Reviews. 57, 2005, S. 411–425. doi:10.1124/pr.57.4.5. Abgerufen am 13. Juni 2011.
- ↑ Josef Platzer, Jutta Engel, Anneliese Schrott-Fischer, Kurt Stephan, Sergio Bova, Howard Chen, Hui Zheng und Joerg Striessnig: Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. In: Cell. 102, 2000, S. 89–97. doi:10.1016/S0092-8674(00)00013-1. Abgerufen am 13. Juni 2011.
- ↑ Sven Moosmang, Verena Schulla, Andrea Welling, Robert Feil, Susanne Feil, Jörg W. Wegener, Franz Hofmann und Norbert Klugbauer: Dominant role of smooth muscle L-type calcium channel Cav1.2 for blood pressure regulation. In: EMBO Journal. 22, 2003, S. 6027–6034. doi:10.1093/emboj/cdg583. Abgerufen am 13. Juni 2011.
- ↑ Nicole Langwieser, Carl J. Christel, Thomas Kleppisch, Franz Hofmann, Carsten T. Wotjak und Sven Moosmang: Homeostatic switch in Hebbian plasticity and fear learning after sustained loss of Cav1.2 calcium channels. In: The Journal of Neuroscience. 23, 2010, S. 8367–8375. doi:10.1523/JNEUROSCI.4164-08.2010. Abgerufen am 13. Juni 2011.
- ↑ Toni Lemke, Andrea Welling, Carl Johannes Christel, Anne Blaich, Dominik Bernhard, Peter Lenhardt, Franz Hofmann und Sven Moosmang: Unchanged β-adrenergic stimulation of cardiac L-type calcium channels in Cav1.2 phosphorylation site S1928A mutant mice. In: The Journal of Biological Chemistry. 283, 2008, S. 34738–34744. doi:10.1074/jbc.M804981200. Abgerufen am 14. Juni 2011.
- ↑ a b Franz Hofmann: Wirkungen von Pharmaka auf den Organismus: allgemeine Pharmakodynamik. In: K. Aktories, U. Förstermann, F. Hofmann und K. Starke: Allgemeine und spezielle Pharmakologie und Toxikologie. 10. Auflage, München, Elsevier GmbH 2009, Seite 5-24. ISBN 978-3-437-42522-6
- ↑ Franz Hofmann, Martin Biel und U. Benjamin Kaupp: International Union of Pharmacology. LI. Nomenclature and structure-function relationships of cyclic nucleotide-regulated channels. 57, 2005, S. 455–462. doi:10.1124/pr.57.4.8. Abgerufen am 26. Juni 2011.
- ↑ a b c Kimberley B. Craven und William N. Zagotta: CNG and HCN channels: two peas, one pod. In: Annual Review of Physiology. 68, 2006, S. 375–401. doi:10.1146/annurev.physiol.68.040104.134728. Abgerufen am 16. Juni 2011.
- ↑ Martin Biel, Xiangang Zong, Madeleine Distler, Eva Bosse, Norbert Klugbauer, Manabu Murakami, Veit Flockerzi und Franz Hofmann: Another member of the cyclic nucleotide-gated channel family, expressed in testis, kidney, and heart. In: Proceedings of the National Academy of Sciences. 91, 1994, S. 3505–3509. doi:10.1073/pnas.91.9.3505. Abgerufen am 16. Juni 2011.
- ↑ Martin Biel, Mathias Seeliger, Alexander Pfeifer, Konrad Kohler, Andrea Gerstner, Andreas Ludwig, Gesine Jaissle, Sascha Fauser, Erberhart Zrenner und Franz Hofmann: Selective loss of cone function in mice lacking the cyclic nucleotide-gated channel CNG3. In: Proceedings of the National Academy of Sciences. 96, 1999, S. 7553–7557. doi:10.1073/pnas.96.13.7533. Abgerufen am 16. Juni 2011.
- ↑ Andreas Ludwig, Xiangang Zong, Michael Jeglitsch, Franz Hofmann und Martin Biel: A family of hyperpolarization-activated cation channels. In: Nature. 393, 1998, S. 587–591. doi:10.1038/31255. Abgerufen am 26. Juni 2011.
- ↑ Andreas Ludwig, Xiangang Zong, Juliane Stieber, Roger Hullin, Franz Hofmann und Martin Biel: Two pacemaker channels from human heart with profoundly different activation kinetics. In: EMBO Journal. 18, 1999, S. 2323–2329. doi:10.1093/emboj/18.9.2323. Abgerufen am 26. Juni 2011.
- ↑ a b c d e Martin Biel, Christian Wahl-Schott, Stylianos Michalakis und Xiangang Zong: Hyperpolarization-activated cation channels: from genes to function. In: Physiological Reviews. 89, 2009, S. 847–885. doi:10.1152/pysiolrev.00029.2008. Abgerufen am 26. Juni 2011.
- ↑ a b Andreas Ludwig, Thomas Budde, Juliane Stieber, Sven Moosmang, Christian Wahl, Knut Holthoff, Anke Langebartels, Carsten Wotjak, Thomas Munsch, Xiangang Zong, Susanne Feil, Robert Feil, Marike Lancel, Kenneth R. Chien, Arthur Konnerth, Hans-Christian Pape, Martin Biel und Franz Hofmann: Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. In: EMBO Journal. 22, 2003, S. 216–224. doi:10.1093/emboj/cdg032. Abgerufen am 16. Juni 2011.
- ↑ T.J. Feuerstein: Antikonvulsiva, Konvulsiva – Pharmakotherapie der Epilepsien. In: K. Aktories, U. Förstermann, F. Hofmann und K. Starke: Allgemeine und spezielle Pharmakologie und Toxikologie. 10. Auflage, München, Elsevier GmbH 2009, S. 283-293. ISBN 978-3-437-42522-6
- ↑ Juliane Stieber, Stefan Herrmann, Susanne Feil, Jana Löster, Robert Feil, Martin Biel, Franz Hofmann und Andreas Ludwig: The hyperpolarization-activated channel HCN4 is required for the generation of pacemaker action potentials in embryonic heart. In: Proceedings of the National Academy of Sciences. 100, 2003, S. 15235–15240. doi:10.1073/pnas.2434235100. Abgerufen am 16. Juni 2011.
- ↑ Stefan Herrmann, Juliane Stieber, Georg Stöckl, Franz Hofmann und Andreas Ludwig: HCN4 provides a „depolarization reserve“ and is not required for heart rate acceleration in mice. In: EMBO Journal. 26, 2007, S. 4423–4432. doi:10.1038/sj.emboj.7601868. Abgerufen am 27. Juni 2011.
- ↑ W. Kobinger, C. Lillie und L. Pichler: N-allyl-derivative of clonidine, a substance with specifc bradycardic action at a cardiac site. In: Naunyn-Schmiedeberg’s Archives of Pharmacology. 306, 1979, S. 255–262
- ↑ K. Aktories, U. Förstermann, F. Hofmann und K. Starke: Allgemeine und spezielle Pharmakologie und Toxikologie. 10. Auflage, München, Elsevier GmbH 2009. ISBN 978-3-437-42522-6
- ↑ Stefan Offermnanns und Walter Rosenthal (Hrsg.): Encyclopedic Reference of Molecular Pharmacology. Berlin, Springer-Verlag 2003. ISBN 3-540-42843-7
Kategorien:- Mediziner (20. Jahrhundert)
- Mediziner (21. Jahrhundert)
- Pharmakologe
- Mitglied der Leopoldina
- Träger des Bundesverdienstkreuzes 1. Klasse
- Träger des Bayerischen Verdienstordens
- Hochschullehrer (TU München)
- Deutscher
- Geboren 1942
- Mann



Wikimedia Foundation.