- Sprachgen
-
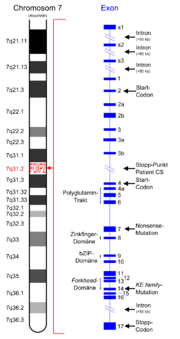 Idiogramm des FOXP2-Gens. Es liegt auf dem q-Arm (langer Arm) von Chromosom 7. Rechts in Blau die 17 Exons von FOXP2.[1]
Idiogramm des FOXP2-Gens. Es liegt auf dem q-Arm (langer Arm) von Chromosom 7. Rechts in Blau die 17 Exons von FOXP2.[1]Das FOXP2-Gen (engl. Abkürzung für Forkhead-Box P2) kodiert das FOXP2-Protein, welches beim Spracherwerb[2], einschließlich grammatikalischer Fähigkeiten, eine große Rolle spielt. Das FOXP2-Protein ist ein Transkriptionsfaktor und zählt zur Gruppe der Forkhead-Box-Proteine. Entdeckt wurde dieses Gen erstmals 1998 bei Untersuchungen einer Londoner Familie, bei der zahlreiche Angehörige unter schweren Sprachstörungen litten. Der Name Forkhead-Box leitet sich von einer bei Taufliegen (Drosophila) beobachteten Mutation (forkhead) ab, die eine gabelartige Veränderung des Kopfes zur Folge hat. Mittlerweile wurde eine Vielzahl von FOX-Genen identifiziert, die in den unterschiedlichsten Organismen zum Teil völlig verschiedene Funktionen haben.
Für das FOXP2-Gen ist in den Massenmedien die Bezeichnung „Sprachgen“ popularisiert worden. Viele andere Wirbeltiere besitzen dieses Gen ebenfalls, und auch in diesen scheint FOXP2 bei der verbalen Kommunikation eine Rolle zu spielen.[3] Nach Ausschaltung des Gens, beispielsweise bei Mäusen (Knockout-Maus) oder durch Mutation beim Menschen, entfaltet es eine pleiotrope Wirkung. Das heißt, dass sich mehrere phänotypische Merkmale ändern. Sprache beruht auf definierten anatomischen, neurologischen, sozialen und psychologischen Grundlagen. Die in der Evolutionslinie zum Homo sapiens stattgefundenen Mutationen haben möglicherweise ausgereicht, um die notwendigen Verknüpfungen im Nervensystem herzustellen, damit der Mensch zu einem Sprachwesen werden konnte.
Inhaltsverzeichnis
Aufbau
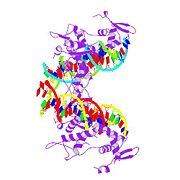 Das vom FOXP2-Gen kodierte FOXP2-Protein. Es besteht aus 715 Aminosäuren
Das vom FOXP2-Gen kodierte FOXP2-Protein. Es besteht aus 715 Aminosäuren Die Struktur des FOXP2-Proteins
Die Struktur des FOXP2-ProteinsDas FOXP2-Gen kodiert das FOXP2-Protein (siehe: Genetischer Code).
FOXP2-Gen
Es wird derzeit geschätzt, dass das FOXP2-Gen mindestens 280 kb (=280.000 Basenpaare) auf Chromosom 7 belegt.[4] Andere Veröffentlichungen von 2007 geben die Zahl der Basenpaare mit 603 kb an.[5] Unabhängig davon bildet eine hohe Anzahl der Basenpaare Introns, das heißt für die Proteinsynthese funktionslose Genbestandteile. 17 Exons, das heißt für das Protein kodierende Bereiche, wurden lokalisiert.[6]
Der Anteil der 2145 (= 715×3) kodierenden an den insgesamt etwa 280.000 nicht kodierenden Basenpaaren ist mit 0,6 % sehr gering, jedoch nicht außergewöhnlich. Generell schwankt das Verhältnis zwischen Introns und Exons von Gen zu Gen sehr stark. So gibt es einige Gene ohne Introns, während andere zu über 95 % aus Introns bestehen.[7]
FOXP2-Protein
Das vom FOXP2-Gen kodierte FOXP2-Protein besteht aus 715 Aminosäuren. Es untergliedert sich in vier Hauptbereiche:
- eine an Polyglutamin reiche Region, die aus zwei benachbarten Polyglutamin-Gebieten besteht und durch sich wiederholende CAG- und CAA-Sequenzen kodiert ist,[8]
- eine Zinkfingerdomäne,
- eine bZIP-Domäne („Leucin-Zipper“) und
- einer Forkhead-Domäne, die aus den Aminosäuren 508 bis 584 gebildet wird.[9]
Die Forkhead-Domäne bindet an DNA. Die Zinkfinger- und die bZIP-Domäne sind für Protein–Protein-Interaktionen wichtig und ebenfalls an der DNA-Bindung beteiligt.[10]
Funktion
Als Transkriptionsfaktor reguliert FOXP2-Protein nach Schätzungen bis zu 1000 andere Gene, welche dies sind, ist jedoch noch weitgehend unbekannt. Über die Auswirkungen eines Ausfalls von FOXP2 liegen dagegen fundierte Kenntnisse vor.
Bereits im Embryo ist das FOXP2-Protein zu finden. Es ist vor allem in den Bereichen exprimiert, aus denen sich später das Kleinhirn (Cerebellum), der Thalamus und die Basalganglien entwickeln. Das Kleinhirn und die Basalganglien spielen beim Erlernen komplexer motorischer Fähigkeiten eine wichtige Rolle. Das Sprechen ist eine Fähigkeit, zu der der Mensch eine komplexe Motorik mühsam erlernen muss.[11]
Sprach- und Sprechstörungen
Das FOXP2-Gen hat bei der Entwicklung der Sprach- und Sprechfähigkeit eine zentrale Funktion. Deshalb führen Mutationen im Gen und ein damit verbundener Funktionsausfall des Proteins zu einer spezifischen Sprach- und Sprechstörung beim Menschen, insbesondere bei der Artikulation und dem Sprachverständnis.[12] Eine Reihe bekannter Sprech- und Sprachstörungen, sowie Autismus, wird daher dem Bereich des FOXP2-Gens auf dem Chromosom 7 zugeordnet.[13][14]
Schizophrenie
Sprachstörungen sind eines der Hauptsymptome einer Schizophrenie. Aus diesem Grund bestand unmittelbar nach der Entdeckung des FOXP2-Gens der Verdacht, dass dieses Gen für die Anfälligkeit zur Ausbildung einer Schizophrenie eine gewisse Rolle spielen könnte. In einer vergleichenden Studie wurden 186 Patienten mit einer Schizophrenie (nach DSM-IV, sie hörten fremde Stimmen) und 160 gesunde Probanden untersucht. Dabei wurden speziell Nukleotidpolymorphismen des FOXP2-Gens analysiert. Es konnten dabei statistisch signifikante Unterschiede bezüglich des Genotypus (P=0,007) und der Allelfrequenzen (P=0,0027) zwischen schizophrenen Patienten mit Gehörhalluzinationen und der Kontrollgruppe, in der einzelnen Nukleotidpolymorphismus rs2396753 gefunden werden. Das Ergebnis lässt den Schluss zu, dass das FOXP2-Gen einen Einfluss auf die Ausbildung einer Schizophrenie haben kann.[15]
Entdeckung
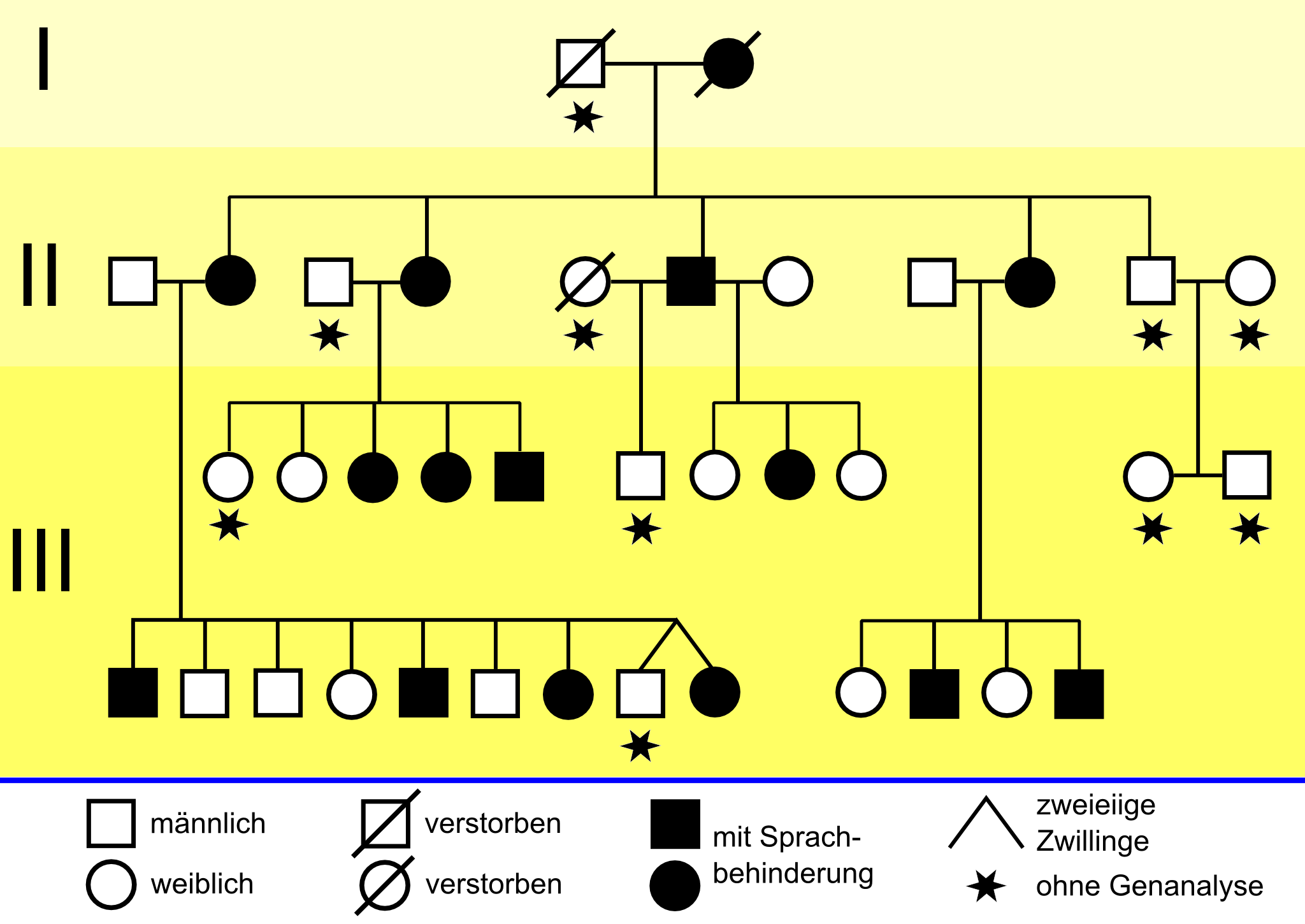
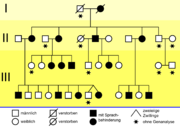 Der Stammbaum der KE-Familie[16]
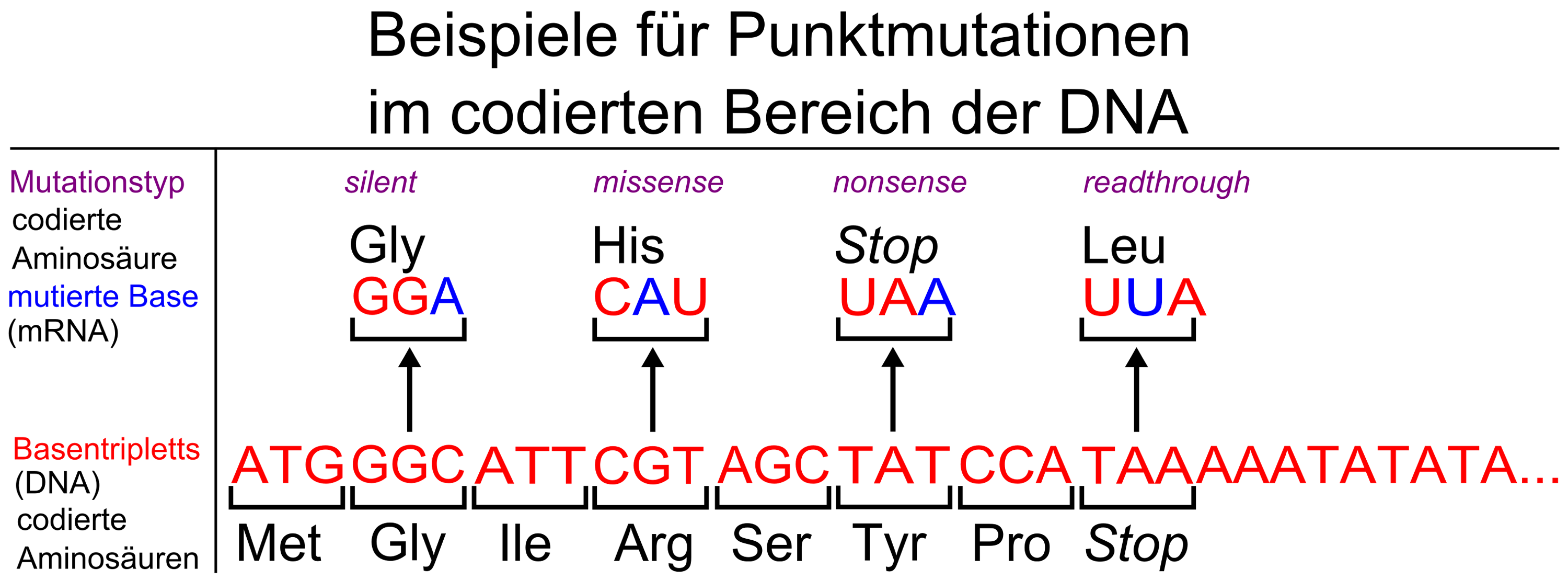
Der Stammbaum der KE-Familie[16]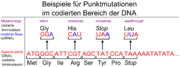 silent-, missense-, nonsense- und readthrough-Mutationen
silent-, missense-, nonsense- und readthrough-Mutationen1990 beschrieben britische Genetiker vom Institute of Child Health eine erbliche Sprachstörung, die drei Generationen einer Familie betrifft. Etwa die Hälfte der 30 Familienmitglieder hat erhebliche Probleme mit Grammatik, Satzbau und Wortschatz.[17] In der wissenschaftlichen Literatur wird diese Gruppe als KE family (KE-Familie) bezeichnet. Sie lebt in Süd-London und ist pakistanischer Herkunft. Der britische Genetiker Anthony Monaco von der Universität Oxford entdeckte 1998 mit seiner Arbeitsgruppe bei den von der Sprachstörung betroffenen Familienmitgliedern auf dem Chromosom 7 einen Abschnitt, den er mit den Sprachproblemen der Familie in Verbindung brachte. Durch genetische Untersuchungen der KE-Familie und eines Jungen (Patient CS), der mit der KE-Familie nicht verwandt ist, aber dennoch die gleichen Symptome aufweist, konnte das sogenannte „Sprachgen“ FOXP2 erstmals identifiziert werden.
Die Mutation des FOXP2-Gens trat offensichtlich erstmals bei der Großmutter der Familie auf. Ihre Sprachstörungen sind so erheblich, dass selbst ihr Ehemann ihre Sätze nur mit Mühe enträtseln kann. Alle drei Töchter[16] und einer ihrer beiden Söhne haben ebenfalls Sprachschwierigkeiten. Von den 24 Enkelkindern zeigen zehn die gleichen Symptome. Die anderen Mitglieder der Familie aus dem Londoner Süden haben keinerlei Verständigungsprobleme.[18] Die bei den betroffenen Mitgliedern der KE-Familie manifestierte Störung wird als verbale Entwicklungsdyspraxie (engl. Developmental Verbal Dyspraxia oder abgekürzt DVD) bezeichnet und unter dem ICD-10 Code F83 („Kombinierte umschriebene Entwicklungsstörungen“) eingeordnet. Eine passende deutsche Umschreibung ist Unfähigkeit zum artikulierten Sprechen.
Symptome bei einer FOXP2-Mutation
Verbale Entwicklungsdyspraxie
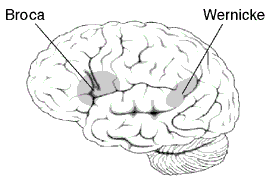
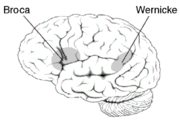 Die Lage der Broca- und Wernicke-Areal im Cortex
Die Lage der Broca- und Wernicke-Areal im CortexDer allgemeine Verhaltensphänotyp, der von der verbalen Entwicklungsdyspraxie betroffenen Personen, zeigt sich in einfachen Wort-Wiederholungstests. Dabei müssen Wörter (beispielsweise: Mörder) und Nichtwörter (beispielsweise: Redröm) nach Vorsagen wiederholt werden. Die von der Mutation betroffen Probanden haben dabei signifikant größere Probleme bei der Artikulation, als die nicht betroffenen.[19] Die Beeinträchtigung erhöht sich stufenweise mit der Kompliziertheit der zu artikulierenden Wörter.
Orofaziale Dyspraxie
Die betroffenen Personen haben auch Schwierigkeiten in der willkürlichen Steuerung der Gesichtsmuskulatur; ein Symptom das orofaziale Dyspraxie genannt wird. Diese Schwierigkeiten können nicht einer allgemeinen Beeinträchtigung der Motorik zugeschrieben werden, da die motorische Leistung der Extremitäten der Patienten von normalen Einzelpersonen nicht zu unterscheiden ist.[19] Auch das Hörvermögen der Patienten ist normal ausgebildet. Der DVD-Phänotyp ähnelt dem, der bei Patienten mit einer Broca-Aphasie beobachtet wird.[20] Allerdings gibt es zwischen beiden Pathologien wichtige Verhaltensunterschiede. So sind Aphasiker im Wortwiederholungstest deutlich besser als bei der Nichtwort-Wiederholung. Die von der FOXP2-Mutation betroffenen Mitglieder der KE-Familie sind dagegen in beiden Tests gleichmäßig schlecht. Eine mögliche Erklärung hierfür ist, dass Aphasiker vor dem Ausbruch ihrer Erkrankung die Assoziation von Lautbildungsmustern zu den entsprechenden Wortbedeutungen erlernt haben. Im Gegensatz dazu hatten die betroffenen Mitglieder der KE-Familie nie die Möglichkeit Wortartikulationsmuster zu erlernen. Deshalb scheitern sie zwangsläufig daran, mit den Wortbedeutungen die Aufgaben des Wort-Wiederholungstest zu lösen.[21]
Weitere Symptome
Zusätzlich zur verbalen und orofazialen Dyspraxie schneiden die von der Mutation betroffenen Mitglieder der KE-Familie bei Tests, die die rezeptive Sprache (das Verständnis von Sprache) und die grammatische Sprache ermitteln, erheblich schlechter als ihre nicht betroffenen Verwandten ab. Das Defizit schließt die Unfähigkeit ein, Wörter richtig zu beugen oder Sätze zu bilden, um einfache Beziehungen von Objekten zu den entsprechenden Abbildungen herzustellen. Zudem zeigen die betroffenen Probanden in nicht-verbalen Intelligenztests eine signifikant niedrigere Intelligenz (IQ-Durchschnittswert: 86, Bereich: 71–111), als die nicht betroffenen (IQ-Durchschnittswert: 104, Bereich: 84–119). Dabei gibt es eine erhebliche Überschneidung zwischen den beiden Gruppen.[19][21][22][23]
Auswirkungen von Veränderungen an FOXP2
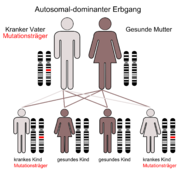 Der autosomal-dominante Erbgang
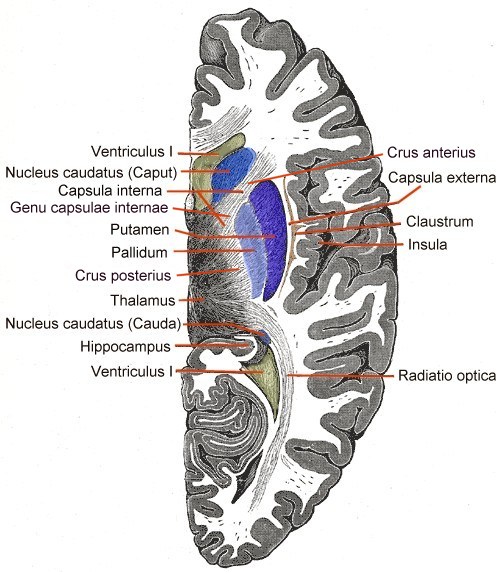
Der autosomal-dominante Erbgang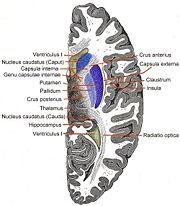 Horizontalschnitt durch das Vorderhirn. In Blau die Basalganglien.
Horizontalschnitt durch das Vorderhirn. In Blau die Basalganglien.Die durch die Mutation bedingten Störungen werden autosomal dominant vererbt. Das FOXP2-Gen befindet sich auf dem langen Arm (q-Arm) von Chromosom 7 in der Bande 7q31. Das Gen wurde ursprünglich, als nur das betroffene Chromosom identifiziert werden konnte, „SPCH1“ (von Speech 1) genannt.[16]
Bei einem 2006 durchgeführten Screening von 49 Probanden die an einer verbalen Dyspraxie leiden, wurde bei einem Probanden eine mütterlicherseits vererbte nonsense-Mutation und bei zwei Probanden eine vermeintlich missense-Mutation des FOXP2-Gens festgestellt.[9] Diese Ergebnisse lassen den Schluss zu, dass FOXP2-Mutationen eine relativ seltene Ursache für Sprech- und Sprachstörungen sind.[1]
Exon 14 (KE-Familie)
Die von der Erbkrankheit betroffenen Mitglieder der KE-Familie haben eine missense-Punktmutation im Exon 14 des Gens. Die Nukleinbase Guanin ist bei ihnen an einer Stelle durch Adenin ersetzt. Dadurch wird in Position 553 des FOXP2-Proteins statt der Aminosäure Arginin die Aminosäure Histidin eingebaut. Im Einbuchstabencode ist die Bezeichnung für Arginin R und für Histidin H. Die Mutation trägt daher die Bezeichnung R553H. Das gebildete Protein kann in Folge des Austausches der Aminosäure seine Funktionen nicht mehr wahrnehmen.
Mittels bildgebender Verfahren des Gehirnes von Mitgliedern der KE-Familie wurden Abnormitäten im Nucleus caudatus, einem Bestandteil der Basalganglien, festgestellt. Erste Einblicke in die neuralen Grundlagen konnten mit Hilfe der funktionellen Magnetresonanztomographie des Gehirnes gewonnen werden. Die von der Mutation betroffenen Mitglieder der KE-Familie wiesen bilaterale strukturelle Defizite auf. Diese zeigten sich vor allem in einer reduzierten Dichte der Grauen Substanz („grauen Zellen“) im Bereich des Nucleus caudatus der Basalganglien[19][23], des vorderen Kleinhirns[24] und des Broca-Areals.[21] Dagegen wurde bei diesen Patienten eine abnormal hohe Dichte an Grauer Substanz im Putamen und im Wernicke-Areal gefunden. Interessanterweise korreliert das Volumen des Nucleus caudatus sehr gut mit der im Sprachtest gezeigten Leistung.[19] Dies ist ein Indiz für den Einfluss des Nucleus caudatus auf die Pathologie der verbalen Entwicklungsdyspraxie (DVD).
Die Basalganglien spielen bei der Bewegungsplanung und -sequenzierung eine entscheidende Rolle.[25] Strukturelle Abweichungen in den striatalen Regionen der Basalganglien (Nucleus caudates und Putamen) bedeuten daher im Allgemeinen eine Beeinträchtigung der Kontrolle über die orofaziale Motorik (Mundmotorik). Unklar ist dagegen, weshalb spezifisch die Mundmotorik beeinträchtigt ist, ohne dass andere motorische Funktionen davon betroffen sind.[21]
Exon 7 (nonsense-Mutation)
2005 wurde an Kindern die nicht zur KE-Familie gehören, aber auch an einer verbalen Dyspraxie leiden, eine sogenannte nonsense-Mutation ebenfalls im FOXP2-Gen entdeckt. Eine nonsense-Mutation ist eine sinnentstellende Mutation, bei der ein Stopcodon entsteht, also ein Basentriplett, das zu einem Abbruch der Synthese des Proteins an dieser Stelle führt. Auch in diesen Fällen mit nonsense-Mutation lassen sich die Sprach- und Sprechdefizite auf die Mutation zurückführen.[9]
Bruchpunkt zwischen Exon 3b und 4
Ein Patient, er wird in der Fachliteratur als Patient CS bezeichnet, hat eine balancierte Translokation zwischen jeweils einer Kopie der Chromosomen 5 und 7 (t(5;7)(q22;q31.2)). Der Bruchpunkt auf Chromosom 7 liegt dabei im FOXP2-Gen zwischen den Exons 3b und 4 und betrifft somit alle bekannten Isoformen des FOXP2-Proteins.[1] Auch dieser Patient leidet an ähnlichen Symptomen wie die von der Mutation betroffenen Mitglieder der KE-Familie.[26]
Gendeletion
Bei einem kanadischen Mädchen wurde 2006 ein Verlust (Deletion) von Teilen des Chromosoms 7 in den Banden 7q31 und 7q32 festgestellt. Im fehlenden Bereich liegt auch das FOXP2-Gen. Das Kind hat schwerwiegende Kommunikationsstörungen in Form einer orofazialen Dyspraxie, eine deutliche Deformität und einen Rückstand in ihrer allgemeinen Entwicklung. Sie kann weder husten noch niesen oder spontan lachen.[27]
Bei Mäusen
 Zwei Knockout-Mäuse
Zwei Knockout-MäuseBei Mäusen wurden im Tierversuch die beiden Exons 12 und 13 von FOXP2 ausgeschaltet („Knockout“). Wenn beide Kopien der FOXP2-Gene unterbrochen wurden, führte dies zu schwerwiegenden Störungen der Motorik, vorzeitigem Tod und einem Ausbleiben der Verständigung mittels Ultraschall. Letztere lässt sich normalerweise dadurch auslösen, dass die Jungtiere von ihrer Mutter entfernt werden. Wurde dagegen nur eines der beiden FOXP2-Gen unterbrochen, so bewirkte dies eine geringe Verzögerung in der Entwicklung der Tiere und eine signifikante Veränderung der Kommunikation mittels Ultraschall. An den Tieren wurden abnorme Veränderungen am Kleinhirn, speziell der Purkinje-Zellen – das sind die Ganglienzellen des Stratum gangliosum der Gyri cerebellares –, festgestellt.[28][29]
Bei Zebrafinken
 Zebrafink (Taeniopygia guttata)
Zebrafink (Taeniopygia guttata)Der Spracherwerb ist nicht auf Menschen beschränkt. Einige Tierarten, darunter Wale, Fledermäuse und Vögel aus drei Ordnungen können ihre akustische Kommunikation („Tiersprache“) durch Imitation erlernen.[21] Singvögel kommunizieren über einen Gesang, den sie weitgehend erlernen müssen. Sie erwerben ihre Lautfolgen dadurch, dass sie ältere Artgenossen imitieren. Von Artgenossen isoliert aufgezogene Jungtiere bleiben dagegen stumm.[11] Singvögel eignen sich daher als Tiermodell für Untersuchungen des Spracherwerbs und seiner genetischen Prädisposition. Bei vielen anderen Tierarten sind dagegen die Lautäußerungen angeboren. Selbst bei Affen geht man davon aus, dass ihr Repertoire an Lauten angeboren ist.[11]
Der Gesang des Zebrafinken (Taeniopygia guttata) besteht aus unterschiedlichen Silben, die strukturierte Reihenfolgen ergeben. Der beim Menschen für den Spracherwerb wichtige Teil des Gehirns liegt in den Basalganglien. Bei Singvögeln wird dieser Bereich Area X genannt. Die Expression von FOXP2 im Area X ist während der Gesangslernphase beim Zebrafinken am höchsten. Bei Kanarienvögeln ist FOXP2 dagegen saisonal unterschiedlich exprimiert. In Phasen, in denen der Gesang umgestellt wird, ist es besonders stark exprimiert. Bei Vogelarten, die ihren Gesang nicht erlernen, wie beispielsweise Ringeltauben, konnten vergleichbare Veränderungen der FOXP2-Expression nicht festgestellt werden.[30]
Mit Hilfe der RNA-Interferenz schalteten Mitarbeiter des Max-Planck-Instituts für molekulare Genetik in Berlin-Dahlem das FOXP2-Gen im Area X von Zebrafinken aus. Bei diesem Verfahren werden kurze komplementäre RNA-Abschnitte in die Zellen eingeschleust, wo sie die mRNA abfangen und die Produktion des FOXP2-Proteins unterdrücken. Die Zebrafinken, bei denen FOXP2 abgeschaltet wurde, imitierten die Silben ihrer älteren Artgenossen weniger präzise und ließen beim Gesang ganze Silben aus.
Der genaue Wirkungsmechanismus von FOXP2 ist noch nicht bekannt.[31] Prinzipiell kann der Gendefekt die motorischen Funktionen, beispielsweise des Stimmkopfs, oder das Abspeichern der zu lernenden Gesänge beeinträchtigen.[32][33]
Die molekulare Evolution von FOXP2
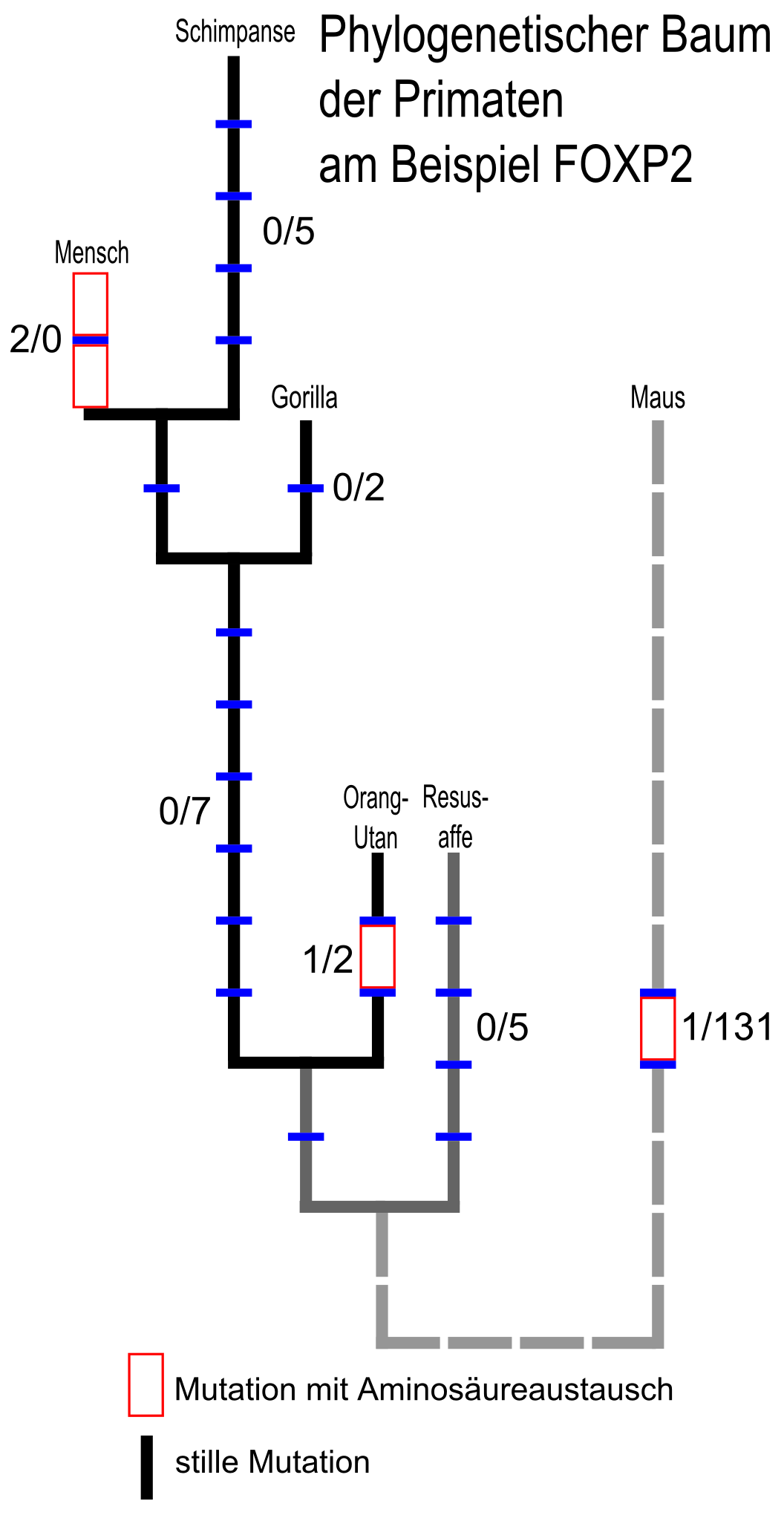
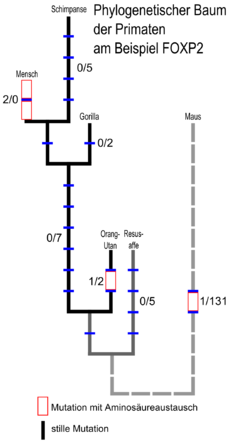 Stammbaum von FOXP2 bei Primaten mit der Maus als Vergleichsgruppe. Die erste Ziffer eines Zahlenpaares gibt die Anzahl der veränderten Aminosäuren (missense-Mutationen), die zweite die Anzahl der stillen Mutationen an. Bei stillen Mutationen ändert sich zwar die Basenabfolge auf der DNA, es wird aber die gleiche Aminosäure codiert (siehe Wobble-Hypothese). Da stille Mutationen deswegen einem erheblich geringeren Selektionsdruck unterliegen, sind sie in der Regel häufiger als missense-Mutationen. Nicht jedoch in der Entwicklungslinie des Menschen. [8]
Stammbaum von FOXP2 bei Primaten mit der Maus als Vergleichsgruppe. Die erste Ziffer eines Zahlenpaares gibt die Anzahl der veränderten Aminosäuren (missense-Mutationen), die zweite die Anzahl der stillen Mutationen an. Bei stillen Mutationen ändert sich zwar die Basenabfolge auf der DNA, es wird aber die gleiche Aminosäure codiert (siehe Wobble-Hypothese). Da stille Mutationen deswegen einem erheblich geringeren Selektionsdruck unterliegen, sind sie in der Regel häufiger als missense-Mutationen. Nicht jedoch in der Entwicklungslinie des Menschen. [8]FOXP2 bei Säuge- und anderen Wirbeltieren
Das FOXP2-Protein gehört bei Säugetieren zu den stark konservierten Proteinen, es unterscheidet sich bei den einzelnen Spezies nur sehr geringfügig.[8] Eine Ausnahme bilden verschiedene Familien der Fledertiere. Dort wurden erhebliche Unterschiede in der FOXP2-Sequenz festgestellt.[34] Dagegen lassen sich nahezu gleiche FOXP2-Proteine beispielsweise bei Singvögeln, Fischen und Reptilien finden.[35][36]
Genabschnitte, die für Polyglutamin kodieren sind generell bekannt dafür, dass sie relativ hohe Mutationsraten aufweisen. Dies ist auch bei den beiden Polyglutamin-Regionen des FOXP2-Gens der Fall. So wiesen alle untersuchten Taxa unterschiedliche Polyglutamin-Längen auf. Für die Funktion des FOXP2-Proteins spielt der Polyglutamin-Bereich eine sehr untergeordnete Rolle. Wenn man diese Regionen außer Acht lässt, unterscheidet sich das menschliche FOXP2-Protein von dem Ortholog der Maus nur in drei Aminosäuren.
Die Evolutionslinien, die zu Mensch und Maus führen, teilten sich vor etwa 70 Millionen Jahren.[37][38] Der letzte gemeinsame Vorfahre von Schimpanse und Mensch lebte vor 4,6 bis 6,2 Millionen Jahren.[39] Von den drei Aminosäure-Unterschieden zwischen Mensch und Maus entstand einer in einem Vorfahren der Maus, keiner zwischen der Trennung der Maus- und Primatenvorfahren bis zur Trennung von Mensch und Schimpanse, und zwei danach (siehe auch Abbildung rechts). Das FOXP2-Protein des Orang-Utan unterscheidet sich durch zwei Aminosäuren von dem der Maus und durch drei vom humanen. Auch beim FOXP2-Protein des Zebrafinken gibt es verglichen mit dem Menschen nur sieben Aminosäure-Unterschiede.[8][40][41]
Die Fähigkeit des Menschen sprechen zu können beruht auf anatomischen und feinmotorischen Fähigkeiten, welche andere Primaten als nächste Verwandte des Menschen nicht aufweisen.[42] Einige Forschungsgruppen nehmen an, dass der Unterschied der beiden Aminosäuren zwischen Schimpanse und Mensch zur Sprachentwicklung beim Menschen geführt hat.[43] Diese These ist jedoch umstritten, da andere Arbeitsgruppen keinen Zusammenhang zwischen Spezies mit erlernter Vokalisierung und solchen mit ähnlicher Mutation in FOXP2 feststellen konnten.[35][36]
Die beiden Unterschiede zu den nächsten Verwandten des Menschen befinden sich im Exon 7. In Position 303 ist dabei Threonin gegen Asparagin und in Position 325 Asparagin gegen Serin ausgetauscht. Die wahrscheinlichen Proteinstrukturen wurden mittels Simulationsrechnungen ermittelt. Die Mutation in Position 325 erzeugt beim humanen FOXP2-Protein eine potenziell reaktive Stelle für die Phosphorylierung durch Proteinkinase C, zusammen mit einer kleinen Veränderung in der Sekundärstruktur des Proteins. Aus verschiedenen Studien ist bekannt, dass die Phosphorylierung von Transkriptionsfaktoren mit Forkhead-Struktur ein wichtiger Mechanismus bei der Genregulation sein kann.[44][45] Zur Klärung, ob die beiden in Exon 7 kodierten Aminosäuren beim Menschen polymorph sind, wurde dieses Exon bei 44 Personen von verschiedenen Kontinenten sequenziert. In keinem Fall wurde eine Form von Aminosäuren-Polymorphismus gefunden.[8]
Fledertiere
 Fledermäuse, im Bild ein Townsend-Langohr (Corynorhinus townsendii), haben die Fähigkeit der Echoortung
Fledermäuse, im Bild ein Townsend-Langohr (Corynorhinus townsendii), haben die Fähigkeit der Echoortung Flughunde, im Bild ein Goldkronen-Flughund (Acerodon jubatus), sind nicht zur Echoortung befähigt
Flughunde, im Bild ein Goldkronen-Flughund (Acerodon jubatus), sind nicht zur Echoortung befähigtWährend bei den meisten Säugetieren durch systematische DNA-Sequenzierungen eine ausgesprochen geringe – nur wenige Aminosäuren betreffende – Mutationsrate auf dem FOXP2-Gen festgestellt werden konnte, sind bei einigen Arten der Fledertiere erhebliche Unterschiede ermittelt worden. Die Fledertiere gehören zu den wenigen Wirbeltieren, die die Fähigkeit besitzen, Laute zu erlernen.[46][47]
Die Ordnung der Fledertiere (Chiroptera) besteht aus zwei Unterordnungen: den Flughunden (Megachiroptera) und den Fledermäusen (Microchiroptera). Fledermäuse nutzen die Echoortung zur Orientierung und zum Beutefang. Bei ihnen sind die sensomotorischen Fähigkeiten besonders hoch entwickelt. Der Empfang der ausgesendeten Ultraschall-Laute erfordert eine ausgeprägte aurale (das Gehör betreffende) und – je nach Fledermausart – orofaziale (Mund) oder nasofaciale (Nase) Abstimmung.[48] Flughunde verfügen dagegen nicht über die Fähigkeit der Echoortung.
Bei der DNA-Sequenzierung wurden die beiden Exons 7 und 17 als die Bereiche identifiziert, in denen – je nach Fledertierart – die größte Variabilität des FOXP2-Gens vorlag. Dabei zeigten sich erhebliche Unterschiede zwischen der FOXP2-Struktur der Fledermäuse und der Flughunde. Die Daten lassen den Schluss zu, dass die Veränderungen des FOXP2-Gens bei Fledermäusen für die Entwicklung der Echoortung eine entscheidende Rolle gespielt haben.[34]
FOXP2 in der Paläogenetik
 Rekonstruktion eines Neandertalerkindes am Anthropologischen Institut der Universität Zürich
Rekonstruktion eines Neandertalerkindes am Anthropologischen Institut der Universität ZürichMit Hilfe der Paläogenetik wurde zunächst berechnet, dass die heute beim Menschen verbreitete Genvariante von FOXP2 zwischen 100.000 und maximal 200.000 Jahre alt sei. Dieser Zeitraum wurde in einem mathematischen Modell, bei dem speziell die Mutationen an Introns betrachtet wurden, ermittelt. Introns sind für die Proteinsynthese funktionslose Genbestandteile. Da sie keine direkte Bedeutung für die Struktur der Proteine haben, ist bei ihnen eine erheblich höhere Mutationsrate als bei Exons zu beobachten. Aus dieser Mutationsrate lässt sich die Geschichte eines Gens rekonstruieren.[49] Der berechnete Zeitraum würde sich mit dem von Paläoanthropologen datierten „Geburtstag“ der Spezies Mensch gut decken.[50][51] Evolutionsgeschichtlich ist dies deutlich später als der ebenfalls per Paläogenetik ermittelte Zeitpunkt der Aufspaltung des evolutionären Stammbaums zwischen Homo sapiens und Homo neanderthalensis vor etwa 300.000 bis 400.000 Jahren. Aus diesen Daten wurde deshalb zunächst geschlossen, dass der Neandertaler nicht über die menschliche Sprachfähigkeit verfügte.[52]
Einige Anthropologen vertreten die Meinung, dass die schnelle Verbreitung des für den Spracherwerb so elementaren FOXP2-Gens für die These spricht, dass Sprache die treibende Kraft bei der Ausbreitung des Menschen auf der Erde war.[53][54]
Die These über das Alter der heutigen FOXP2-Genvariante des Menschen und daraus abgeleitet, dass der Neandertaler deshalb nicht über die menschliche Sprachfähigkeit verfügte, musste im Oktober 2007 revidiert werden. Aus Neandertaler-Knochen wurde das FOXP2-Gen sequenziert. Dabei wurde kein Unterschied in der Sequenz des Neandertalers im Vergleich zum modernen Menschen gefunden.[55][56]
Die DNA-Sequenzierung prähistorischer Funde ist ein sehr aufwändiges Verfahren. Die Proben enthalten nur sehr geringe Mengen endogener DNA. Zudem ist die Kontaminierung der Proben und Reagenzien[57] mit menschlicher DNA ein erhebliches Problem, vor allem dadurch, dass sich die DNA des Neandertalers nur sehr wenig von der des modernen Menschen unterscheidet.[58] Aus zwei unterschiedlichen Neandertaler-Knochen, die 2006 in der asturischen El Sidrón-Höhle gefunden wurden[59], wurde zunächst die mitochondriale DNA (mtDNA) der ca. 43.000 Jahre alten Proben analysiert. Anhand der mtDNA lässt sich durch einige bekannte Substitutionen feststellen, ob es sich um die DNA eines modernen Menschen oder eines Neandertalers handelt.[60] Nachdem feststand, dass bei den Proben eindeutig die DNA eines Neandertalers vorlag, wurden auf Exon 7 des FOXP2-Gens die beiden Bereiche untersucht, die als Mutationen seit der Aufspaltung in Menschen und Schimpansen bekannt sind. Dabei wurde kein Unterschied in der entsprechenden Sequenz des Neandertalers und der des modernen Menschen festgestellt. Der Neandertaler verfügte somit ebenfalls über die die Sprache ermöglichende Mutation des FOXP2-Gen.[56] Die Möglichkeit, dass sich das Gen durch gemeinsame Kinder von Homo sapiens und Homo neanderthalensis sowohl beim modernen Menschen als auch bei den Neandertalern durchsetzte, wird anhand von Ergebnissen aus Studien an mitochondrialer DNA ausgeschlossen.[61]
Literatur
Fachartikel
- C. S. Lai et al.: A forkhead-domain gene is mutated in a severe speech and language disorder. In: Nature, 413/2001, S. 519–23. PMID 11586359
- F. Vargha-Khadem et al.: FOXP2 and the Neuroanatomy of Speech and Language. In: Nature Reviews Neuroscience, 6/2005, S. 131–138. PMID 15685218
- F. Vargha-Khadem et al.: Praxic and nonverbal cognitive deficits in a large family with a genetically transmitted speech and language disorder. In: Proc Nat Acad Sci, 92/1995, S. 930–33. PMID 7846081
- H. A. Bruce, R. L. Margolis: FOXP2: novel exons, splice variants, and CAG repeat length stability. In: Human Genetics, 111/2002, S. 136–44. PMID 12189486
- C. S. Lai u. a.: A novel forkhead-domain gene is mutated in a severe speech and language disorder. In: Nature, 413/2001, S. 519–23. PMID 11586359
- B. Wang u. a.: Multiple domains define the expression and regulatory properties of Foxp1 forkhead transcriptional repressors. In: J Biol Chem, 278/2003, S. 24259–68. PMID 12692134
- J. C. Stroud u. a.: Structure of the forkhead domain of FOXP2 bound to DNA. In: Structure, 14/2006, S. 159–66. PMID 16407075
Lehrbücher und Hochschulschriften
- W. Bigenzahn, G. Böhme: Sprach-, Sprech-, Stimm- und Schluckstörungen. Elsevier Deutschland, ISBN 3-437-46950-9
- R. J. McCauley: Assessment of Language Disorders in Children., Lawrence Erlbaum Associates, 2001, S. 118, ISBN 0-8058-2562-2
- S. Otte: Gibt es Zusammenhänge zwischen einer expressiven Sprachentwicklungsstörung und einem zentro- temporalen Sharp- Wave Fokus (Rolando- Fokus) mit der weiteren Entwicklung? Dissertation, 2005, Julius-Maximilians Universität zu Würzburg S. 82–83.
- H. Teepe: Welche Bedeutung haben die Neurowissenschaften für die Fremdsprachendidaktik?, Dissertation, RWTH Aachen, 2005
Populärwissenschaftlich
- U. Bahnsen, U. Willmann: Wie Gene die Lippen spitzen. In: Die Zeit, 21/2001.
- R. Berger: Warum der Mensch spricht: Eine Naturgeschichte der Sprache. Eichborn 2008.
- S. Haesler: Also sprach der Zebrafink. In: Gehirn&Geist, 12/2006, S. 52–57. (pdf-Datei)
- A. Leßmöllmann: Raus mit der Sprache! In: Zeit Wissen, 1/2006.
- B. P. Trivedi: Scientists Identify a Language Gene. In: National Geographic Today, 4. Oktober 2001 (englisch)
Weblinks
- FOXP2 in der RCSB Protein-Datenbank (englisch)
- K. Seefeldt: Der kleine Unterschied. Telepolis vom 16. August 2002
- U. Bahnsen: Sprich oder stirb! Sonntagszeitung vom 18. August 2002
- A. MacAndrew: FOXP2 and the Evolution of Language (englisch)
Einzelnachweise
- ↑ a b c L. Feuk u. a.: Absence of a Paternally Inherited FOXP2 Gene in Developmental Verbal Dyspraxia. In: Am J Hum Genet., 79/2006, S. 965–72. PMID 17033973
- ↑ C. Lai u. a.:The SPCH1 region on human 7q31: genomic characterization of the critical interval and localization of translocations associated with speech and language disorder. In: Am J Hum Genet, 67/2000, S. 357–68. PMID 10880297
- ↑ P. Schlobinski: Grammatikmodelle: Positionen und Perspektiven,Vandenhoeck & Ruprecht, 2003, S. 83–84, ISBN 3-525-26530-1
- ↑ Ensembl.org: Ensembl Gene Report for ENSG00000128573 eingesehen am 2. August 2008
- ↑ A. F. Wright, N. Hastie: Genes and Common Diseases: Genetics in Modern Medicine.,Cambridge University Press, 2007, ISBN 0-521-83339-6
- ↑ J. Zhang u. a.: Accelerated Protein Evolution and Origins of Human-Specific Features: FOXP2 as an Example. In: Genetics, 162/2002, S. 1825–35. PMID 12524352
- ↑ E. H. McConekey: How the Human Genome works., Jones & Bartlett, 2004, ISBN 0-7637-2384-3 S. 5
- ↑ a b c d e W. Enard u. a.: Molecular evolution of FOXP2, a gene involved in speech and language. In: Nature, 418/2002, S. 869–872. PMID 12192408 doi:doi:10.1038/nature01025
- ↑ a b c K. D. MacDermot u. a.: Identification of FOXP2 Truncation as a Novel Cause of Developmental Speech and Language Deficits. In: Am J Hum Genet, 76/2005, S. 1074–80. PMID 15877281
- ↑ U. Wahn (Hrsg.): Pädiatrische Allergologie und Immunologie, Elsevier Deutschland, S. 895, ISBN 3-437-21311-3.
- ↑ a b c S. Haesler: Also sprach der Zebrafink. In: Gehirn&Geist, 12/2006, S. 52–57.
- ↑ Presseinformation der Max-Planck-Gesellschaft: Geschwätzige Zebrafinken, vom 31. März 2004
- ↑ E. K. O’Brien u. a.: Association of Specific Language Impairment (SLI) to the Region of 7q31. In: Am. J. Hum. Genet., 72/2003, S. 1536–43. PMID 12721956
- ↑ T. H. Wassink u. a.: Evaluation of FOXP2 as an autism susceptibility gene. In: American Journal of Medical Genetics, 114/2002, S. 566–9. PMID 12116195
- ↑ J. Sanjuán u. a.: Association between FOXP2 polymorphisms and schizophrenia with auditory hallucinations. In: Psychiatr Genet, 16/2006, S. 67–72. PMID 16538183
- ↑ a b c S. E. Fisher u. a.: Localisation of a gene implicated in a severe speech and language disorder. In: Nature Genetics 18/1998, S. 168–70. PMID 9462748
- ↑ J. Cohen: Die Evolution der Sprache. In: Technology Review,2/2008
- ↑ U. Bahnsen, U. Willmann: Wie Gene die Lippen spitzen. In: Die Zeit, Ausgabe 51/2001
- ↑ a b c d e K. E. Watkins u. a.: Behavioural analysis of an inherited speech and language disorder: comparison with acquired aphasia. In: Brain, 125/2002, S. 452–64. PMID 11872604
- ↑ A. R. Damasio, N. Geschwind: The neural basis of language. In: Annu Rev Neurosci, 7/1984, S. 127–47.
- ↑ a b c d e S. Haesler: Studien zur Evolution und Funktion des FoxP2-Gens in Singvögeln., Dissertation, FU Berlin, 2007
- ↑ K. J. Alcock u. a.: Oral dyspraxia in inherited speech and language impairment and acquired dysphasia. In: Brain Lang, 75/2000, S. 17–33. PMID 11023636
- ↑ a b F. Vargha-Khadem u. a.: Neural basis of an inherited speech and language disorder. In: Proc Natl Acad Sci U S A, 95/1998, S. 12695–700. PMID 9770548
- ↑ E. Belton u. a.: Bilateral brain abnormalities associated with dominantly inherited verbal and orofacial dyspraxia. In: Hum Brain Mapp, 18/2003, S. 194–200. PMID 12599277
- ↑ A. M. Graybiel: Building action repertoires: memory and learning functions of the basal ganglia. In: Curr Opin Neurobiol., 5/1995, S. 733–41. PMID 8805417
- ↑ P. Markl: Warum sprechen Menschen? In Wiener Zeitung, Ausgabe vom 13. September 2002
- ↑ S. Zeesman u. a.: Speech and language impairment and oromotor dyspraxia due to deletion of 7q31 that involves FOXP2. In: Am J Med Genet A, 140/2006, S. 509–14. PMID 16470794
- ↑ W. Shu u. a.: Altered ultrasonic vocalization in mice with a disruption in the Foxp2 gene. In: Proc Natl Acad Sci U S A., 102/2005, S. 9643–8. PMID 15983371
- ↑ E. Fujita u. a.: Ultrasonic vocalization impairment of Foxp2 (R552H) knockin mice related to speech-language disorder and abnormality of Purkinje cells. In: Proc Natl Acad Sci U S A, 105/2008, S. 3117–22. PMID 18287060
- ↑ S. Haesler u. a.: FoxP2 expression in avian vocal learners and non-learners. In: J Neurosci, 24/2004, S. 3164–75. PMID 15056696
- ↑ S. A. White u. a.: Singing Mice, Songbirds, and More: Models for FOXP2 Function and Dysfunction in Human Speech and Language. In: The Journal of Neuroscience, 26/2006, S. 10376–9. PMID 17035521
- ↑ Max-Planck-Gesellschaft: Schlechte Gesangsschüler – Wissenschaftler schalten das Gen FOXP2 bei Zebrafinken stumm und kriegen was zu hören. Pressemitteilung vom 4. Dezember 2007
- ↑ S. Haesler u. a.: Incomplete and inaccurate vocal imitation after knockdown of FoxP2 in songbird basal ganglia nucleus Area X. In: PLoS Biol., 5/2007, e321. PMID 18052609
- ↑ a b G. Li u. a.: Accelerated FoxP2 evolution in echolocating bats. In: PLoS ONE, 2/2007, e900. PMID 17878935
- ↑ a b D.M. Webb, J. Zhang: FoxP2 in song-learning birds and vocal-learning mammals. In: J Hered., 96/2005, S. 212–6. PMID 15618302
- ↑ a b C. Scharff, S. Haesler: An evolutionary perspective on FoxP2: strictly for the birds? In: Curr Opin Neurobiol, 15/2004, S. 694–703. PMID 16266802
- ↑ S. Kumar, S. B. Hedges: A molecular timescale for vertebrate evolution. In: Nature, 392/1998, S. 917–20. PMID 9582070
- ↑ E. Eizirik u. a.: Molecular dating and biogeography of the early placental mammal radiation. In: J. Hered., 92/2001, S. 212–19. PMID 11396581
- ↑ F. C. Chen, W. H. Li: Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. In: Am. J.Hum. Genet., 68/2001, S. 444–56. PMID 11170892
- ↑ I. Teramitsu u. a.: Parallel FoxP1 and FoxP2 expression in songbird and human brain predicts functional interaction. In: J Neurosci., 24/2004, S. 3152–63. PMID 15056695
- ↑ S. Haesler u. a.: FoxP2 expression in avian vocal learners and non-learners.. In: J Neurosci., 24/2004, S. 3164–75. PMID 15056696
- ↑ P. Liebermann: The Biology and Evolution of Language., Harvard University Press, 1984, ISBN 0-674-07413-0
- ↑ W. Enard u.a. Molecular evolution of FOXP2, a gene involved in speech and language. In: Nature, 418/2002, S. 869–72. PMID 12192408
- ↑ G. J. Kops u. a.:Control of cell cycle exit and entry by protein kinase b-regulated forkhead transcription factors. In: Mol. Cell. Biol., 22/2002, S. 2025–36. PMID 11884591
- ↑ A. Brunet u.a. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. In: Cell, 96/1999, S. 857–68. PMID 10102273
- ↑ J. W. Boughman: Vocal learning by greater spear-nosed bats. In: Proc Biol Sci, 265/1998, S. 227–33. PMID 9493408
- ↑ G. Jones, R. D. Ransome: Echolocation calls of bats are influenced by maternal effects and change over a lifetime. In: Proc Biol Sci, 252/1993, S. 125–8. PMID 8391702
- ↑ C. F. Moss CF, S. R. Sinha: Neurobiology of echolocation in bats. In: Current Opinion in Neurobiology, 13/2003, S. 751–8. PMID 14662378
- ↑ Paläogenetik: Wie alt ist die Sprache? In: Geo Magazin, 10/2002
- ↑ U. Bahnsen: Paläogenetik In: Zeit Wissen, 34/2002
- ↑ R. G. Klein: The Human Career: Human Biological and Cultural Origins., University Chicago Press, 1989, ISBN 0-226-43963-1
- ↑ E. D. Jarasch: Genetische Spuren der Menschwerdung, vom 22. Dezember 2006
- ↑ J. Müller-Jung: Frankfurter Allgemeine Zeitung, Ausgabe vom 15. August 2002
- ↑ F. Carmine: Genomtechnologie und Stammzellforschung- ein verantwortbares Risiko? Govi-Verlag Eschborn, 2003, S. 53, ISBN 3-7741-1000-X
- ↑ M. Inman: Neandertals Had Same „Language Gene“ as Modern Humans. In: National Geographic News, 18. Oktober 2007
- ↑ a b J. Krause u. a.: The derived FOXP2 variant of modern humans was shared with Neandertals. In: Current Biology, 17/2007, S. 1908–12. PMID 17949978
- ↑ M. Hofreiter u. a.; Ancient DNA. In: Nat. Rev. Genet., 2/2001, S. 353–9. PMID 11331901
- ↑ S. Pääbo: Human evolution. In: Trends Cell Biol, 9/1999, M13–6. PMID 10611673
- ↑ A. Rosas u. a.: Paleobiology and comparative morphology of a late Neandertal sample from El Sidron, Asturias, Spain. In: Proc. Natl. Acad. Sci. USA, 103/2006, S. 19266–71. PMID 17164326
- ↑ R. E. Green u. a.: Analysis of one million base pairs of Neanderthal DNA. In: Nature, 444/2006, S. 330–6. PMID 17108958
- ↑ Archaeologie-online.de: Neandertaler hat Sprachgen., vom 19. Oktober 2007





Wikimedia Foundation.