- G6PD-Mangel
-
ICD-10-Codes G6PD-Mangel
(Glucose-6-phosphat-Dehydrogenase-Mangel)D55.0 Glucose-6-Phosphat-
Dehydrogenase (G6PD)-Mangel
mit AnämieE74.8 Glucose-6-Phosphat-
Dehydrogenase (G6PD)-Mangel
ohne AnämieInformationen in medizinischen Datenbanken OMIM Datensatz +305900 HGMD Datensatz G6PD Der Glucose-6-Phosphat-Dehydrogenase-Mangel (kurz: G6PD(H)-Mangel; Synonyme: Favismus, Favabohnen-Krankheit) ist ein angeborener Mangel des Enzyms Glucose-6-phosphat-Dehydrogenase beim Menschen durch Mutation des G6PD-Gens auf dem X-Chromosom, Abschnitt q28 (Xq28). Der Mangel des Enzyms G6PD führt durch Veränderung des Zuckerstoffwechsels zu einer vermehrten Zerstörbarkeit der roten Blutkörperchen (Erythrozyten) durch äußere Faktoren in Form einer Hämolyse. Die Symptome des G6PD-Mangels sind je nach Mutation (allelische Variante) und Geschlecht des Betroffenen hoch variabel: sie reichen von Beschwerdefreiheit bis hin zu einer lebensbedrohlichen hämolytischen Krise. Drei typische Formen sind Beschwerdefreiheit, eine induzierte hämolytische Anämie (hämolytische Krise) und eine chronische hämolytische Anämie mit hämolytischen Krisen. Neugeborene mit einem G6PD-Mangel können eine verlängerte oder besonders ausgeprägte Neugeborenengelbsucht erleiden (Ikterus neonatorum). Von den schweren Verlaufsformen sind fast ausschließlich Jungen und Männer betroffen.
Unter Zusammenfassung aller Varianten und Ausprägungen weisen auf der Welt ca. 200 Millionen Menschen irgendeine Form des G6PD-Mangels auf. Vorwiegend wird der G6PD-Mangel in geographischen Regionen angetroffen, in denen Malaria verbreitet war oder ist. In malaria-freien Gebieten kommt der G6PD-Mangel ebenfalls vor, ist aber deutlich seltener. Diese Verteilung des G6PD-Mangels stellt einen balancierten Polymorphismus dar: die relative Resistenz der betroffenen Frauen gegenüber Malaria-Infektionen wiegt unter Gesichtspunkten der Evolution den Nachteil der Empfindlichkeit der betroffenen Männer auf. Die Erkrankung wurde im Zusammenhang mit dem Genuss von dicken Bohnen (Fava-Bohne, Ackerbohne, Saubohne) erstmalig beobachtet, worauf der alternative Name Favismus zurückführbar ist. Neben den Fava-Bohnen besitzen Substanzen wie Henna oder diverse Medikamente die Eigenschaft, hämolytische Krisen auszulösen.
Die Behandlung des G6PD-Mangels richtet sich nach der vorliegenden Form und dem Betroffenen. Für ausnahmslos alle Betroffenen ist das Wissen um den G6PD-Mangel entscheidend, da somit auslösende Faktoren einer hämolytischen Krise wie Fava-Bohnen oder bestimmte Medikamente vermieden werden können. Bei einer hämolytischen Krise sind sofortige Vermeidung oder Entfernung des Auslösers entscheidend; Bluttransfusionen können in seltenen Fällen erforderlich sein. Die Prognose des G6PD-Mangels hängt von der vorhandenen Form des G6PD-Mangels ab: Sie ist insgesamt gut, bei vorhandener Aufklärung der Betroffenen und leichter Form des G6PD-Mangels sehr gut und geht nicht mit einer verminderten Lebenserwartung einher. Menschen mit schwerem G6PD-Mangel und häufigen hämolytischen Krisen haben eine höhere Morbidität (Krankheitsanfälligkeit) als Menschen ohne G6PD-Mangel.
Inhaltsverzeichnis
Ursache
Normale Funktion von G6PD
Die Glucose-6-phosphat-Dehydrogenase (G6PD; EC 1.1.1.49) ist ein Enzym mit einer Molaren Masse von maximal 59.289 Dalton (Da).[1][2] Es umfasst 249 bis 515 Aminosäuren und bildet im Raum ein Homodimer oder Homotetramer. Sie gehört zur Enzymfamilie der Oxidoreduktasen und bildet eine eigene Unterfamilie der Glucose-6-phosphat-Dehydrogenasen. G6PD existiert in 2 Isoformen: eine kurze und eine lange Isoform. Die lange Isoform (515 Aminosäuren) findet sich in Lymphoblasten, Granulozyten und Spermazellen. Die kurze Isoform (249 Aminosäuren) findet sich in der Leber und vor allem in den roten Blutkörperchen (Erythrozyten). Beide Isoformen binden NADP zweifach: einmal am N-terminalen Ende des Enzyms als Kofaktor (zwischen der Aminosäure 27 und 210) und einmal als strukturelles Element am C-terminalen Ende.
Das Enzym G6PD hat die Aufgabe die Reaktion von Glucose-6-Phosphat nach D-Glucono-1,5-Lakton-6-phosphat (6-Phosphoglucono-δ-lakton) unter Umwandlung von NADP+ nach NADPH zu katalysieren. Das im Rahmen der katalysierten Reaktion entstandene NADPH wird durch das Enzym Glutathionreduktase als Kofaktor weiter verwendet. Glutathionreduktase wandelt das oxidierte Tripeptid Glutathion zum reduzierten Glutathion um, wobei NADPH zu NADP+ oxidiert wird. Glutathion ist ein in fast allen Zellen des Menschen vorkommendes Tripeptid, welches als Radikalfänger (Antioxidans) fungiert: aggressive Oxidantien wie beispielsweise Sauerstoff-Radikale, Hydroxyl-Radikale, Wasserstoffperoxid, Primaquin[3][4][5], Nitrofurantoin[6], Sulfanilamid, Vicin und Convicin. Beide zuletzt genannten Substanzen sind Alkaloide der Ackerbohne. Bestimmte Medikamente können ebenfalls Radikale bilden, weisen diese in sich bereits auf oder werden unter Radikalbildung verstoffwechselt. Das reduzierte Glutathion steht der Zelle und dem Organismus im Ganzen im reduzierten Zustand als Antioxidans zur Verfügung, im oxidierten Zustand kann Glutathion die antioxidative Funktion nicht ausüben.
Das bei Reduzierung von Glutathion durch die Glutathionreduktase entstehende NADP+ wird durch die Glucose-6-phosphat-Dehydrogenase wieder zu NADPH umgewandelt: zusammenfassend bewirkt G6PD ein "Recycling" von NADPH, wodurch das Glutathion immer wieder in seinen reduzierten (antioxidativ wirksamen) Zustand gebracht werden kann. Somit ist G6PD für die Aufrechterhaltung der antioxidative Kapazität im Menschen mit verantwortlich.
Pathomechanismus (Pathophysiologie)
Wenn das Enzym Glucose-6-phosphat-Dehydrogenase durch Veränderungen im G6PD-Gen in seiner Aktivität gemindert oder weniger als normal gebildet oder gar nicht mehr gebildet, bewirken diese Veränderungen eine Verminderung der Umwandlung von NADP nach NADPH. Gleichzeitig vermindert sich auch die Umwandlung von Glucose-6-Phosphat nach 6-Phosphoglucono-δ-lacton. Die verminderte Umwandlung von NADP in NADPH zieht aber Konsequenzen im Glutathionstoffwechsel nach sich. Das Enzym Glutathionreduktase, dass für die Reduktion von oxidiertem Glutathion ohne antioxidative Wirkung in das reduzierte Glutathion mit antioxidativer Wirkung unter gleichzeitiger Transformation von NADPH nach NADP verantwortlich ist, kann aufgrund des Mangels an NADPH diese Reaktion nicht mehr wie gewöhnlich katalysieren. Als Folge dieser verminderten oder ausgefallenen Reduktion von oxidiertem Glutathion sinkt die Menge an reduziertem Glutathion (antioxidative Wirkung) ab. Damit ist die antioxidative Kapazität des menschlichen Körpers vermindert.
Werden nun Substanzen mit oxidativer Wirkung (Oxidantien) dem Körper zugeführt, kann bei stark abgesunkener Menge an reduziertem Glutathion die antioxidative Schutzwirkung nicht aufrecht erhalten werden: durch die Oxidantien kommt es zu Schädigungen der Zellbestandteile, insbesondere der Zellmembran, aber auch an Eiweißen. Eine Fortsetzung dieser oxidativen Schädigungen führt letztendlich zur unumkehrbaren Schädigung der betroffenen Zellen und nachfolgend zu ihrem Untergang.
Im Gegensatz zu anderen Zellen des menschlichen Körpers beziehen Erythrozyten ihre Energie fast ausschließlich aus Glucose. 90-95% der Glucose werden über die Glykolyse zur Gewinnung von ATP und damit Energie benutzt. Die verbleibenden 5-10% Glucose werden durch den Pentosephosphat-Zyklus (Hexosemonophosphat-Shunt) zur Bildung von NADPH durch die Glucose-6-phosphat-Dehydrogenase verwendet. Andere Mechanismen zur NADPH-Gewinnung liegen im Erythrozyten in nicht nennenswertem Anteil vor. Ist die Aktivität der Glucose-6-phosphat-Dehydrogenase vermindert oder fällt ganz aus, sinkt die Menge an NADPH, nachfolgend die Menge an reduziertem Glutathion. Somit verfügt der Erythrozyt de facto über keine antioxidativen Schutzmechanismen mehr. Bei Einwirkung von Oxidantien bei Menschen mit G6PD-Mangel werden die Erythrozyten geschädigt und anschließend zerstört. Diese Zerstörung der Erythrozyten in Blutgefäßen ist eine Form der Hämolyse. Tritt diese schnell ablaufend auf, so entspricht dies einer hämolytischen Krise mit nachfolgender schwerer hämolytischer Anämie. Bei langsamen Ablauf entsteht eine hämolytische Anämie leichterer Ausprägung.
Da im menschlichen Organismus allein durch die Verstoffwechselung von Sauerstoff (Erythrozyten transportieren Sauerstoff mittels Bindung an Hämoglobin) zu jedem Zeitpunkt Sauerstoff-Radikale anfallen, sind die Erythrozyten bei einem G6PD-Mangel immer betroffen. Je nach Genmutation und nachfolgend Veränderung des Enzyms G6PD herrscht entweder eine Hämolyse variablen Ausmaßes vor oder die Lebensdauer der Erythrozyten ist verkürzt. Kompensatorisch wird die Bildung roter Blutkörperchen (Erythropoese) gesteigert, wobei der Kompensation Grenzen gesetzt sind.
Der kürzere Lebensdauer der Erythrozyten bei G6PD-Mangel wird auch für die relative schützende Wirkung eines G6PD-Mangels gegen eine Infektion mit Plasmodium falciparum (Malaria) verantwortlich gemacht. Die in den Erythrozyten lebenden Plasmodien finden durch den gesteigerten Umsatz von Erythrozyten (vermehrte Zerstörung alter Erythrozyten und vermehrte Produktion junger Erythrozyten) weniger Möglichkeiten eines parasitischen Befalls.
Vererbung
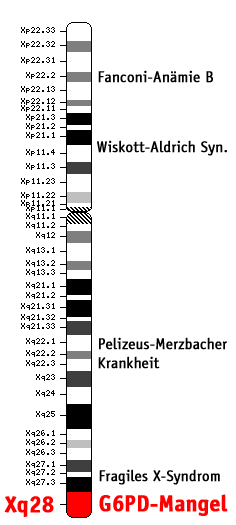
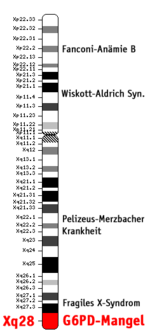 Bild 3. Position des G6PD-Gens auf menschlichem X-Chromosom
Bild 3. Position des G6PD-Gens auf menschlichem X-Chromosom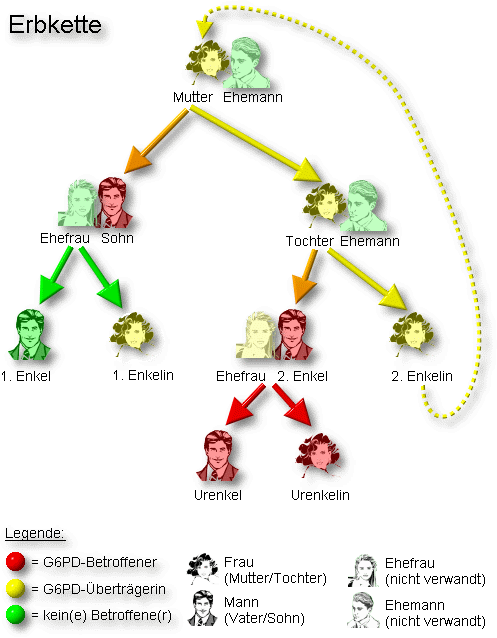 Bild 4. Erbgang (Biologie) des G6PD-Mangels (genetische Übertragung mit allen Kombinationen)
Bild 4. Erbgang (Biologie) des G6PD-Mangels (genetische Übertragung mit allen Kombinationen)Das Glucose-6-phosphat-Dehydrogenase-Gen (G6PD-Gen) befindet sich auf Abschnitt q28 des X-Chromosoms (Xq28) beim Menschen (DEHUG6 Locus; siehe Bild 3). Daher erfolgt die Vererbung des G6PD-Gens mit dem X-Chromosom (X-chromosomal; siehe Bild 4).
Wird ein verändertes oder krankhaftes G6PD-Gen auf einem X-Chromosom an die Nachkommenschaft (Kinder) weitergegeben, so ist aufgrund der X-chromosomalen Vererbung das Geschlecht der nachkommenden Betroffenen von besonderer Bedeutung. Bei einem weiblichen Nachkommen und Weitergabe eines mutierten G6PD-Gens steht auf dem zweiten X-Chromosom ein gesundes G6PD-Gen zur Verfügung (Heterozygotie). Entsprechend der X-Inaktivierung (Lyon-Hypothese) werden zufällig und ungeordnet (randomisiert) G6PD-Gene mitsamt dem X-Chromosom aktiviert oder deaktiviert. Das deaktivierte X-Chromosom mit dem damit deaktivierten G6PD-Gen kann nicht als Grundlage für die Proteinproduktion fungieren. Entsprechend werden mittels inaktiven defekten G6PD-Gene keine defekten G6PD-Enzyme produziert. Intakte inaktive G6PD-Gene werden ebenfalls nicht zur Synthese von G6PD-Enzym verwendet. Es resultieren bei den so betroffenen weiblichen Individuen zwei Gruppen von Erythrozyten - eine mit verändertem G6PD-Gen und eine mit normalem G6PD-Gen. Je nach Verteilung der beiden Gruppen, die entsprechend der zufällig ablaufenden X-Inaktivierung sehr unterschiedlich ausfallen kann, resultiert bei betroffenen Mädchen und Frauen eine hoch variable und zumeist schwache Merkmalsausprägung des G6PD-Mangels und somit - ebenfalls sehr variabel - wenig bis keine Krankheitszeichen.
Bei männlichen Nachfahren, denen ein X-Chromosom mit Defekten des G6PD-Gens vererbt wird, fehlt das zweite X-Chromosom. Sie sind für die Erkrankung hemizygot. Dies bedingt auch das Ausbleiben der X-Inaktivierung, so dass die Erbinformationen des einzigen X-Chromosoms immer in Proteine übersetzt werden. Bei Vorhandensein eines defekten G6PD-Gens wird somit immer ein entsprechend defektes G6PD-Enzym produziert. Da das Y-Chromosom keine Informationen über G6PD enthält, entsteht bei männlichen Nachkommen immer nur eine Gruppe von Erythrozyten mit defektem G6PD-Enzym. Als Folge dessen weisen betroffene Jungen und Männer in aller Regel eine erheblich schwerere Symptomatik als betroffene Mädchen oder Frauen auf, da sie über keine Gruppe von roten Blutkörperchen mit normaler G6PD-Aktivität verfügen. Die Wahrscheinlichkeit an einem schwerwiegendem G6PD-Mangel zu erkranken ist für Jungen und Männer somit massiv höher als für Mädchen oder Frauen.
Mädchen oder Frauen erkranken nur dann schwerer an einem G6PD-Mangel, wenn beide X-Chromosomen jeweils ein defektes G6PD-Gen aufweisen (homozygot). Dies setzt zwingend voraus, dass sowohl der Vater als auch die Mutter solcher weiblicher Nachkommen eine Mutation des G6PD-Gens aufweisen müssen. Dies ist selten der Fall.
Varianten des G6PD-Gens
Das Enzym G6PD weist zwei Varianten auf: A und B. Diese Varianten werden durch genetische Änderungen auf DNA-Ebene bedingt. Die resultierenden Eiweisse haben im Gegensatz zu den Mutationen keine krankhafte Funktion. Die A-Variante wird durch Austausch eines DNA-Bausteins (Nukleotid) an Position 376 der DNA des G6PD-Gens definiert (Nukleotidsubstitution). Für das das Nukleotid Adenin kommt Guanin zum Einsatz. Dieser Tausch bewirkt eine Änderung der durch diesen DNA-Abschnitt verschlüsselten Aminosäure. Anstelle der Asparaginsäure wird Asparagin verschlüsselt, so dass das resultierende G6PD-Protein entsprechend verändert ist. Die B-Variante hat diese Änderung der DNA-Bausteinabfolge (DNA-Basensequenz) an der Position 376 nicht. Ein wesentlicher Unterschied in der Funktion und Funktionsfähigkeit der beiden Varianten des G6PD-Enzyms besteht ohne das Vorliegen weiterer Veränderungen (Mutationen) nicht.
Die Varianten des G6PD-Enzym haben eine bestimmte geographische Verteilung. Die A-Variante findet sich vorwiegend bei Menschen aus Afrika (alle Regionen unterhalb der Sahara) und bei Menschen mit afrikanischem Ursprung wie die afro-amerikanische Bevölkerungsgruppe in den USA. Auch in China ist die A-Variante sehr verbreitet. Die B-Variante ist für den Mittelmeerraum typisch. Sie kommt sowohl in südeuropäischen Bevölkerungsgruppen wie auch in nahöstlichen und nordafrikanischen Bevölkerungsgruppen vor.
Menschen mit der A-Variante von G6PD weisen im Falle einer Mutation des G6PD-Gens als grobe Regel ausgedrückt weniger schwere Krankheitszeichen auf als Menschen mit der B-Variante. Es ist aber durchaus möglich und vorkommend, dass Menschen mit einer krankhaften A-Variante der G6PD schwerere Erkrankungszeichen haben als Menschen mit einer krankhaften B-Variante.
Mutationen des G6PD-Gens
Das G6PD-Gen hat eine Größe von ca. 18.000 Basenpaaren (bp) und weist 13 Exons auf. Die Exons kodieren für das bis 249-515 Aminosäuren umfassende Protein Glucose-6-phosphat-Dehydrogenase (je nach Isoform). Insgesamt sind gegenwärtig (23. September 2006) 149 Mutationen des G6PD-Gens bekannt, molekulargenetisch identifiziert und in der Fachliteratur beschrieben. Davon sind 139 Missense- oder Nonsense-Mutationen, 8 kleine Deletionen, 1 große Deletion und 1 Mutation des Splicings.
Es sind folgende Varianten und Mutationen des G6PD-Gens bekannt und molekulargenetisch beschrieben:
-
Tabelle 1. Beschriebene Mutationen und Varianten der G6PD - Auswahl Variante oder Mutation G6PD Gen Protein Bezeichnung Kurzname Isoform
G6PD-ProteinOMIM-Code Typ Untertyp Position Position Strukturänderung Funktionsänderung G6PD-A(+) Gd-A(+) G6PD A +305900.0001 Polymorphismus Nukleotid A→G 376
(Exon 5)126 Asparagin→Asparaginsäure (ASN126ASP) Kein Enzymdefekt (Variante) G6PD-A(-) Gd-A(-) G6PD A +305900.0002 Substitution Nukleotid G→A 376
(Exon 5)
und
20268
und
126Valin→Methionin (VAL68MET)
Asparagin→Asparaginsäure (ASN126ASP)G6PD-Mediterran Gd-Med G6PD B +305900.0006 Substitution Nukleotid C→T 563
(Exon 6)188 Serin→Phenylalanin (SER188PHE) Klasse II G6PD-Canton Gd-Canton G6PD A +305900.0021 Substitution Nukleotid G→T 1376 459 Arginin→Leucin (ARG459LEU) Klasse II G6PD-Chatham Gd-Chatham G6PD +305900.0003 Substitution Nukleotid G→A 1003 335 Alanin→Threonin (ALA335THR) Klasse II G6PD-Cosenza Gd-Cosenza G6PD B +305900.0059 Substitution Nukleotid G→A 1376 459 Arginin→Prolin (ARG459PRO) G6PD-Aktivität <10%, somit hoher Anteil von Kranken. G6PD-Mahidol Gd-Mahidol G6PD +305900.0005 Substitution Nukleotid G→A 487
(Exon 6)163 Glycin→Serin (GLY163SER) Klasse II G6PD-Orissa Gd-Orissa G6PD +305900.0047 Substitution Nukleotid 44 Alanin→Glycin (ALA44GLY) NADP-Bindungsstelle betroffen. Höhere Stabilität als andere Varianten. G6PD-Asahi Gd-Asahi G6PD A- +305900.0054 Substitution Nukleotid (mehrere) A→G
±
G→A376
(Exon 5)
202126
68Asparagin→Asparaginsäure (ASN126ASP)
Valin→Methionin (VAL68MET)Klasse III.
Epidemiologie (Vorkommen)
Ca. 200 Millionen Menschen sind vom G6PD-Mangel in irgendeiner Form weltweit betroffen. Dabei sind Menschen aus gegenwärtigen oder ehemaligen Malaria-Gebieten deutlich häufiger vom G6PD-Mangel in allen Formen betroffen als Menschen, die nicht aus solchen Regionen stammen. Entsprechend dem Ausbreitungsgebiet der Malaria (sowohl gegenwärtiges als auch vergangenes) finden sich vermehrte Häufigkeiten des G6PD-Mangels in den Tropen und Subtropen. Folgende Regionen sind durch ein vermehrtes Vorkommen des G6PD-Mangels gekennzeichnet:
-
Tabelle 2. Prävalenz und Inzidenz des G6PD-Mangels nach geographischen Regionen der Erde - Auswahl Nordamerika Hier sind die Bevölkerungsgruppen mit afro-amerikanischen oder mittelmeer-europäischen wie auch nahöstlichen und fernöstlichen Abstammungshintergrund betroffen USA In der afro-amerikanischen Bevölkerungsgruppe weisen 12,8% aller Neugeborenen dieser Gruppe einen G6PD-Mangel auf. [7] Mittelamerika Alle Bevölkerungsgruppen betroffen Mexiko Prävalenz aller G6PD-Formen 0,71%. [8] Südamerika Alle Bevölkerungsgruppen betroffen Brasilien Im Bundesstaat Rio Grande do Sul, Neugeborene (n=2799): 1,4% vollständiger G6PD-Mangel, 6,4% teilweise G6PD-Mangel, gesamt 7,9%. [9] Naher Osten Alle Bevölkerungsgruppen betroffen. Israel 806 männliche und weibliche Neugeborene wurden untersucht: 30,2% aller Jungen und 10,4% aller Mädchen hatten einen schweren G6PD-Mangel. 14% der betroffenen Mädchen hatten einen Vater, welcher aus einer Population mit niedriger Prävalenz für den G6PD-Mangel stammt. [10] Libanon Betroffene Männer 10:1.000, betroffene Frauen 0,4:1.000. Bei Männern über 14 Jahre 36 Betroffene in 3.000 untersuchten Personen (1,2% oder kumulative Inzidenz 12:1.000). [11] Jordanien Neugeborenen-Screening bei 181 Neugeborenen. 11% aller weiblichen und 12% aller männlichen Neugeborenen weisen eine Form des G6PD-Mangels auf. [12] Zypern 6,4% der erwachsenen Männer haben einen G6PD-Mangel: davon haben 52,6% die mediterrane Mutation 563C->T im Exon 6 (Ser188Phe) des G6PD-Gens. [13] Südeuropa Alle Bevölkerungsgruppen betroffen. Griechenland 1.286.000 Neugeborene (männlich und weiblich) wurden zwischen 1977 und 1989 untersucht. Ein G6PD-Mangel kam bei 3,14% aller Untersuchten vor (entspricht 40.349 Betroffene); 1 aus 22 männlichen und 1 aus 54 weiblichen Untersuchten war betroffen. [14] Italien| Im Neugeborenen-Screening Inzidenz durch Messungen 0,9:1.000, kalkuliert nach Hardy-Weinberg-Gesetz 4,8:1.000. [15] In der Provinz Cosenza wurde bei 209 von 16.787 Jungen ein G6PD-Mangel vorgefunden (1,24%). 99 der 209 betroffenen Jungen hatten einen schweren G6PD-Mangel (0,59%). [16] Portugal 0,51% von 15.208 Männern haben einen G6PD-Mangel. [17] Spanien Neugeborenen-Screening auf G6PD-Mangel in Katalonien: von 3.189 untersuchten Neugeborenen (inklusive Neugeborene mit Migranten-Hintergrund) wiesen 29 eine Form des G6PD-Mangels auf (0,91%), wobei 3 von 29 erfassten Fällen bei Neugeborenen spanischer Herkunft vorzufinden waren. [18] In Menorca beträgt die Prävalenz 9,7/1.000 bei Männern (11 Betroffene aus 1139 Untersuchten, entsprechend 0,97%). Nordafrika Alle Bevölkerungsgruppen betroffen. Libyen Bei Männern beträgt die Prävalenz des G6PD-Mangels in Ost-Libyen 2,8%, bei Frauen 1,8%. [19] Tunesien Bei 325 Untersuchten betrug die Inzidenz des G6PD-Mangels 1,84%. Unter den Betroffenen wiesen 96,2% den mediterranen Typ B(+) auf, 1,96% den afrikanischen Typ A(+). [20] Asien - Mittlerer Osten Alle Bevölkerungsgruppen betroffen. Abu Dhabi Neugeborenen-Screening bei 8.198 Neugeborenen: Betroffenen vom G6PD-Mangel sind 9,1% (746) Neugeborene. [21] Bahrain Prävalenz 25% im Neugeborenen-Screening 1985. [22] Irak Iran In den iranischen Provinzen Mazandaran und Guilan am Kaspischen Meer beträgt die Prävalenz des G6PD-Mangels 8,6-16,4%. Folgende Varianten von G6PD wurden dabei vorgefunden: G6PD-Mediterran 66,2%, G6PD-Chatham 27% und G6PD-Cosenza bei 6,75% der Betroffenen. [23][24] Asien - Ferner Osten Alle Bevölkerungsgruppen betroffen. Afghanistan Bei einer Untersuchung an afghanischen Flüchtlingen in Pakistan wiesen 15,8% der Angehörigen der Bevölkerungsgruppe der Pathan und 7,0-9,1% der Usbeken eine Form des G6PD-Mangels auf. 2,9% der Tadschiken und 2,1% der Turkmenen waren ebenfalls betroffen. [25] China In Gesamt-China hatten 6.683 von 155.879 freiwilligen Testpersonen (4,29%) irgendeine Form des G6PD-Mangels. [26] Indien In der Provinz Rajasthan wurde bei 55 von 1.198 Kindern (4,59%) eine Form des G6PD-Mangels festgestellt. [27] Indonesien In Nord-Sumatra wurde bei 6,0% aller Jungen in der Provinz Nias 3,9% aller Jungen der Provinz Asahan und 0,9% aller Jungen in der Stadt Medan eine Form des G6PD-Mangels festgestellt. [28] Japan 9.620 Kinder von Überlebenden der Atombombenabwürfe wurden in Hiroshima und Nagasaki untersucht. 0,11% der männlichen und 0,42% der weiblichen Kinder in Hiroshima sowie 0,16% der männlichen und 0,31% der weiblichen Kinder in Nagasaki hatten eine Form des G6PD-Mangels. Insgesamt wurden 10 Varianten bzw. Veränderungen des G6PD-Enzyms hierbei identifiziert, wobei jeweils 3 neue Varianten in Hiroshima und Nagasaki beschrieben werden konnten. [29] Malaysia Neugeborenen-Screening bei 8.975 Neugeborenen zwischen 1985 und 1986 zeigte in der chinesischen Bevölkerungsgruppe eine Inzidenz von 4,5%, in der malaysischen 3,5% und in der indischen 1,5%. [30] Myanmar (Burma) Im Bundesstaat (Region) Shan mit Malaria-Endemie 66 von 311 Untersuchten mit G6PD-Mangel entsprechend 21,2% (Variante G6PD-Mahdiol). [31] Pakistan Alle Formen des G6PD-Mangels kommen bei 1,8% der männlichen Bevölkerung vor (1,07% in Kaschmiris, 1,47% in bei Bewohnern des Punjab, 2,77% bei Sindhis und 3,17% in Pathanern). [32] Singapur Neugeborenen-Screening von ca. 1.600.000 Neugeborenen mit einer Inzidenz von 1,62% bei allen Neugeborenen (3,15% bei männlichen, 0,11% bei weiblichen). [33] Tadschikistan Prävalenz 2,1% (alle Formen des G6PD-Mangels) [34] Thailand Prävalenz beim männlichen Geschlecht 3 bis 18% in Abhängigkeit von der geographischen Region: die häufigste G6PD-Mutation ist G6PD-Mahidol (163Gly-->Ser). [35] Prävalenz bei 505 untersuchten männlichen Neugeborenen beträgt 12,08%. [36] 61 von 505 männlichen Neugeborenen wiesen eine Form des G6PD-Mangels auf. [37] Vietnam Prävalenz in der Bevölkerungsgruppe der Kinh 0,5% und Mong 0,7%: beide Gruppen leben nicht in Malaria-Endemiegebieten. In der Bevölkerung, welche in Malaria-Endemiegebieten Vietnams lebt, beträgt die Prävalenz 9,7 bis 31%. [38] Afrika (Sub-Sahara) Alle Bevölkerungsgruppen betroffen Kenia Prävalenz im Flachland des Malaria-Endemiegebiets 7%, im Hochland des Malaria-Endemiegebiets 1%. [39] Südafrika In der Bevölkerungsgruppe mit griechischer Abstammung in Kapstadt konnte bei 6,7% aller männlichen Untersuchten eine Form des G6PD-Mangels festgestellt werden. [40] Nord- und Mitteleuropa Bevölkerungsgruppen mit Migrationshintergrund aus subtropischen und tropischen Regionen betroffen (inkl. Mittelmeerraum) Frankreich Im Neugeborenen-Screening 6% durch G6PD-Mangel betroffene Jungen und 1% betroffene Mädchen. [41] Niederlande 668 schwangere Frauen und 754 gesunde Neugeborene ethnischer Minderheiten in den Niederlanden wurden auf das Vorliegen eines G6PD-Mangels untersucht. Die Prävalenz über alle Gruppen zusammengefasst betrug 6,6% bei Männern und 5,2% bei Frauen. Das höchste Vorkommen des G6PD-Mangels fand sich bei Afrikanern sub-saharischer Herkunft (Schwarzafrikanern). [42]
Symptome und klinische Formen
Der Glucose-6-phosphat-Dehydrogenase-Mangel verläuft aufgrund seiner unterschiedlichen Formen und Ausprägungen sehr unterschiedlich. Von Bedeutung für das Vorhandensein von Symptomen beziehungsweise die klinische Form sind neben der genetischen Veränderung im G6PD-Gen und dem Geschlecht auch das mögliche Vorhandensein anderer angeborener oder erworbener Bluterkrankungen (Beispiel: Eisenmangelanämie) sowie die Exposition des Betroffenen gegenüber Substanzen, welche eine Hämolyse auslösen können.
Beschwerdefreiheit
Die meisten Mädchen und Frauen mit einem G6PD-Mangel sind im Kindes- und Erwachsenenalter beschwerdefrei. Zwar kann bei diesen Patienten oder Betroffenen mittels Aktivitätsmessung der G6PD in den Erythrozyten eine Aktivitätsminderung festgestellt werden; zu einer Hämolyse mit nachfolgenden klinischen Symptomen wie Gelbsucht (Ikterus), Blutarmut (Anämie), Schwächegefühl (Malaise) oder Kreislaufversagen (in schweren Fällen der Hämolyse) kommt es nicht. Auch wenn Substanzen, die eine Hämolyse auslösen können, eingenommen oder zugeführt werden, tritt eine Hämolyse praktisch nie auf, da die Restaktivität von G6PD zur Verhinderung einer Schädigung (Hämolyse) der Erythrozyten fast immer ausreicht.
Lediglich im Neugeborenenalter kann bei Vorliegen eins Glucose-6-phosphat-Dehydrogenase-Mangels eine verlängerte oder vermehrte Neugeborenengelbsucht (Ikterus neonatorum) auftreten, welche allerdings nicht zwingend zu einer Erkrankung oder Schädigung des betroffenen Neugeborenen führen muss. Allein auf Basis der Symptome ist ein G6PD-Mangel bedingter verlängerter oder vermehrter Neugeborenenikterus von einem Neugeborenenikterus anderer Ursache nicht zu unterscheiden.
Hämolytische Krise
Chronisch-hämolytische Anämie
Diagnose
Klassifikation
Entsprechend der gemessenen Funktionsfähigkeit (Enzymaktivität) von G6PD kann der G6PD-Mangel nach der WHO in verschiedene Klassen eingeteilt werden.[43]
-
Tabelle 3. Klassifikation des G6PD-Mangels nach Enzymaktivität - WHO Klassifikation Klasse WHO Enzymaktivität in Erythrozyten in %Normal Klinisches Bild Klasse 1 vermindert chronisch hämolytische Anämie Klasse 2 <10% schwerer G6PD-Mangel Klasse 3 10–60% mäßiger G6PD-Mangel Klasse 4 normale Aktivität (60%-100%) kein G6PD-Mangel Klasse 5 gesteigerte Aktivität (>110%) kein G6PD-Mangel
Differentialdiagnose
Der G6PD-Mangel kann aufgrund seiner variablen Symptome und Ausprägungen teilweise nur sehr schwer gegenüber anderen Erkrankungen abgegrenzt werden.
Gelbsucht (Ikterus)
Zwischen der durch einen G6PD-Mangel verursachten Gelbsucht (Ikterus) und anderen Formen der Gelbsucht, die durch andere Umstände und Auslöser verursacht werden, muss unterschieden werden:
- Neugeborenengelbsucht (Ikterus neonatorum)
- Bei der klassischen Neugeborenengelbsucht fehlen die Symptome und Zeichen einer Hämolyse, welche beim G6PD-Mangel anzutreffen sind, in aller Regel.
- Hämolytischer Ikterus des Neugeborenen (Morbus haemolyticus neonatorum)
- Die Unterscheidung zwischen diesem Ikterus und dem des G6PD-Mangels kann sehr schwer sein. Der Morbus haemolyticus neonatorum wird durch einen positiven Coombs-Test bewiesen, welcher den Nachweis für die hämolyse-verursachenden Antikörper führt. Ein negativer Coombs-Test spricht eher für einen G6PD-Mangel; den letztendlichen Beweis kann nur eine Bestimmung der G6PD-Aktivität in den Erythrozyten bewerkstelligen, wobei insbesondere nach Bluttransfusionen bei schweren hämolytischen Krisen dies sehr problematisch sein kann. Alternativ erfolgt eine Genotypisierung des G6PD-Gens.
Hämolytische Anämien
- Angeborene hämolytische Anämie(n) durch Enzymdefekte
- Neben dem G6PD-Mangel existieren noch andere angeborene hämolytische Anämien, welche durch Enzymdefekte verursacht werden. Ein Beispiel für eine solche Anämie ist der Pyruvatkinase-Mangel (PK-Mangel).
- β-Thalassämie (Beta-Thalassämie)
- β-Thalassämien sind ebenfalls angeborene hämolytische Anämien. Im Gegensatz zu den durch Enzymdefekten hervorgerufenen Anämien haben β-Thalassämien ihre Ursache in einer Mutation der β-Kette des Hämoglobins. Sie sind im Gegensatz zum G6PD-Mangel eine Hämoglobinopathie. Bei typischen β-Thalassämien tritt die hämolytische Anämie erst mit 3-9 Monaten in Erscheinung, wenn durch die Umstellung der Hämoglobinsynthese vom fetalen Hämoglobin (HbF) auf das adulte Hämoglobin (HbA) die Mutation in der beta-Kette des Hämoglobins ihre Auswirkungen zeigen kann. Auch ist der Verlauf einer hämolytischen Anämie infolge β-Thalassämie meistens gradueller als die des G6PD-Mangels.
- α-Thalassämie (alpha-Thalassämie)
- Wie β-Thalassämie.
- Autoimmunhämolytische Anämie(n)
- Bei den unterschiedlichen Formen der autoimmunhämolytischen Anämien lassen sich zumeist die Antikörper, welche zur Hämolyse mittels Immunprozessen führen, identifizieren. Dieser Nachweis von Antikörpern ist beim G6PD-Mangel nicht möglich, da die Hämolyse hier durch einen anderen Mechanismus erfolgt.
- Medikamenten-induzierte hämolytische Anämie(n)
- Diese hämolytischen Anämien können sehr schwer von einem G6PD-Mangel zu unterscheiden sein. Ein Hinweis kann die Herkunft des Betroffenen geben, wobei medikamenten-induzierte hämolytische Anämien auch bei Menschen aus Regionen mit G6PD-Mangel vorkommen können. Die sichere Unterscheidung gelingt erst durch die Messung der G6PD-Aktivität.
Andere Anämien
- Eisenmangelanämie
- Diese Anämie geht typischerweise nicht mit einer Hämolyse einher. In aller Regel entwickelt sie sich auch graduell. Typischerweise finden sich kleine Erythrozyten (MCV kleiner 70 fL) und ein Mangel an Eisen und Ferritin im Serum bzw. Plasma.
- Blutungsanämie
- Diese Anämie kann durch Identifizierung einer Blutungsquelle gegenüber der Anämie des G6PD-Mangels abgegrenzt werden. Ausserdem weist eine Blutungsanämie in aller Regel keine Zeichen einer Hämolyse auf.
- Folsäuremangelanämie
- Diese Anämie ist charakterisiert durch übernormal große Erythrozyten (MCV größer als 100 fL). Sie entwickelt sich graduell und hat normalerweise keinerlei Hämolysezeichen.
- Vitamin B12 Mangelanämie
- Diese Anämie ist charakterisiert durch übernormal große Erythrozyten (MCV größer als 100 fL). Sie entwickelt sich graduell und hat normalerweise keinerlei Hämolysezeichen.
Therapie
Aufklärung
Vermeidung hämolytischer Krisen
Behandlung hämolytischer Krisen
G6PD-Mangel und Malaria
Historisches
Der Mangel an Glucose-6-phosphat-Dehydrogenase als Ursache des bereits länger bekannten Krankheitsbild des Favismus beziehungsweise der hämolytischen Anämie infolge Primaquin-Exposition wurden 1956 erstmalig identifiziert.[44] Die erste cDNA-Sequenz der G6PD wurde 1981 beschrieben[45]
Quellen
Lehrbücher
- Ohls RK, Christensen RK: Chapter 20. Diseases of the blood. In: Behrman RE, Kliegman RM, Jenson HB: Nelson Textbook of Pediatrics. 17th edition. Saunders, Philadelphia. Mai 2003. ISBN 0-7216-9556-6
- Recht M, Pearson HA: Chapter 295. Hemolytic Anemias. In: McMillan JA, Deangelis CD, Feigin RD, Warshaw JB, Oski FA: Oski's Pediatrics: Principles and Practice. 3rd Edition. Lippincott Williams & Wilkins, . Juni 1999. ISBN 0-7817-1618-7
Übersichtsarbeiten
Originalarbeiten
- ↑ Takizawa T, Huang IY, Ikuta T, Yoshida A. Human glucose-6-phosphate dehydrogenase: primary structure and cDNA cloning. Proc Natl Acad Sci U S A 1986; 83(12):4157-4161.
- ↑ Martini G, Toniolo D, Vulliamy T et al. Structural analysis of the X-linked gene encoding human glucose 6-phosphate dehydrogenase. EMBO J 1986; 5(8):1849-1855.
- ↑ Dern RJ, Weinstein IM, Leroy GV, Talmage DW, Alving AS. The hemolytic effect of primaquine. I. The localization of the drug-induced hemolytic defect in primaquine-sensitive individuals. J Lab Clin Med 1954; 43(2):303-309. PMID 13130947
- ↑ Dern RJ, Beutler E, Alving AS. The hemolytic effect of primaquine. II. The natural course of the hemolytic anemia and the mechanism of its self-limited character. J Lab Clin Med 1954; 44(2):171-176. PMID 13184224
- ↑ Beutler E, Dern RJ, Alving AS. The hemolytic effect of primaquine. III. A study of primaquine-sensitive erythrocytes. J Lab Clin Med 1954; 44(2):177-184. PMID 13184225
- ↑ Kimbro EL Jr, Sachs MV, Torbert JV. Mechanism of the hemolytic anemia induced by nitrofurantoin (furadantin); further observations on the incidence and significance of primaquine-sensitive red cells. Bull Johns Hopkins Hosp 1957; 101(5):245-257. PMID 13472250
- ↑ Kaplan M, Herschel M, Hammerman C, Hoyer JD, Stevenson DK. Hyperbilirubinemia among African American, glucose-6-phosphate dehydrogenase-deficient neonates. Pediatrics 2004; 114(2):e213-e219.
- ↑ Vaca G, Arambula E, Esparza A. Molecular heterogeneity of glucose-6-phosphate dehydrogenase deficiency in Mexico: overall results of a 7-year project. Blood Cells Mol Dis 2002; 28(3):436-444.
- ↑ Castro S, Weber R, Dadalt V, Tavares V, Giugliani R. Prevalence of G6PD deficiency in newborns in the south of Brazil. J Med Screen 2006; 13(2):85-86.
- ↑ Kaplan M, Hammerman C, Kvit R, Rudensky B, Abramov A. Neonatal screening for glucose-6-phosphate dehydrogenase deficiency: sex distribution. Arch Dis Child Fetal Neonatal Ed 1994; 71(1):F59-F60.
- ↑ Khneisser I, Adib SM, Loiselet J, Megarbane A. Prevalence of G6PD deficiency and knowledge of diagnosis in a sample of previously unscreened Lebanese males: clinical implications. J Med Screen 2006; 13(1):26-28.
- ↑ Talafih K, Hunaiti AA, Gharaibeh N, Gharaibeh M, Jaradat S. The prevalence of hemoglobin S and glucose-6-phosphate dehydrogenase deficiency in Jordanian newborn. J Obstet Gynaecol Res 1996; 22(5):417-420. PMID 8987321
- ↑ Drousiotou A, Touma EH, Andreou N et al. Molecular characterization of G6PD deficiency in Cyprus. Blood Cells Mol Dis 2004; 33(1):25-30.
- ↑ Missiou-Tsagaraki S. Screening for glucose-6-phosphate dehydrogenase deficiency as a preventive measure: prevalence among 1,286,000 Greek newborn infants. J Pediatr 1991; 119(2):293-299.
- ↑ Zaffanello M, Rugolotto S, Zamboni G, Gaudino R, Tato L. Neonatal screening for glucose-6-phosphate dehydrogenase deficiency fails to detect heterozygote females. Eur J Epidemiol 2004; 19(3):255-257.
- ↑ Tagarelli A, Bastone L, Cittadella R, Calabro V, Bria M, Brancati C. Glucose-6-phosphate dehydrogenase (G6PD) deficiency in southern Italy: a study on the population of the Cosenza province. Gene Geogr 1991; 5(3):141-150. PMID 1841600
- ↑ Martins MC, Olim G, Melo J, Magalhaes HA, Rodrigues MO. Hereditary anaemias in Portugal: epidemiology, public health significance, and control. J Med Genet 1993; 30(3):235-239.
- ↑ Manu-Pereira MM, Maya A, Cararach V et al. [Neonatal screening of hemoglobinopathies and glucose-6-phosphate dehydrogenase in Catalonia. Pilot study in anonymous not related population]. Med Clin (Barc) 2006; 126(8):281-285.
- ↑ Mir NA, Fakhri M, Abdelaziz M et al. Erythrocyte glucose-6-phosphate dehydrogenase status of newborns and adults in eastern Libya. Ann Trop Paediatr 1985; 5(4):211-213. PMID 2418771
- ↑ Blibech R, Gharbi Y, Mrad A et al. Incidence of glucose-6-phosphate dehydrogenase (G6PD) deficiency in Tunisian populations. Nouv Rev Fr Hematol 1989; 31(3):189-191. PMID 2616266
- ↑ Abdulrazzaq YM, Micallef R, Qureshi M et al. Diversity in expression of glucose-6-phosphate dehydrogenase deficiency in females. Clin Genet 1999; 55(1):13-19. PMID 10066026
- ↑ Al AS. Campaign to control genetic blood diseases in Bahrain. Community Genet 2005; 8(1):52-55.
- ↑ Ohkura K, Miyashita T, Nakajima H et al. Distribution of polymorphic traits in Mazandaranian and Guilanian in Iran. Hum Hered 1984; 34(1):27-39. PMID 6234219
- ↑ Mesbah-Namin SA, Sanati MH, Mowjoodi A, Mason PJ, Vulliamy TJ, Noori-Daloii MR. Three major glucose-6-phosphate dehydrogenase-deficient polymorphic variants identified in Mazandaran state of Iran. Br J Haematol 2002; 117(3):763-764. PMID 12028056
- ↑ Bouma MJ, Goris M, Akhtar T, Khan N, Khan N, Kita E. Prevalence and clinical presentation of glucose-6-phosphate dehydrogenase deficiency in Pakistani Pathan and Afghan refugee communities in Pakistan; implications for the use of primaquine in regional malaria control programmes. Trans R Soc Trop Med Hyg 1995; 89(1):62-64. PMID 7747310
- ↑ Jiang W, Yu G, Liu P et al. Structure and function of glucose-6-phosphate dehydrogenase-deficient variants in Chinese population. Hum Genet 2006; 119(5):463-478.
- ↑ Choubisa SL, Choubisa L, Pande S, Srivastava YK. Incidence of abnormal haemoglobins and G-6-PD deficiency in school children of Udaipur (Rajasthan), India. J Trop Med Hyg 1987; 90(4):215-216. PMID 2443725
- ↑ Matsuoka H, Ishii A, Panjaitan W, Sudiranto R. Malaria and glucose-6-phosphate dehydrogenase deficiency in North Sumatra, Indonesia. Southeast Asian J Trop Med Public Health 1986; 17(4):530-536. PMID 3576282
- ↑ Kageoka T, Satoh C, Goriki K et al. Electrophoretic variants of blood proteins in Japanese. IV. Prevalence and enzymologic characteristics of glucose-6-phosphate dehydrogenase variants in Hiroshima and Nagasaki. Hum Genet 1985; 70(2):101-108. PMID 3977788
- ↑ Hon AT, Balakrishnan S, Ahmad Z. Hyperbilirubinaemia and erythrocytic glucose 6 phosphate dehydrogenase deficiency in Malaysian children. Med J Malaysia 1989; 44(1):30-34. PMID 2626111
- ↑ Than AM, Harano T, Harano K, Myint AA, Ogino T, Okadaa S. High incidence of 3-thalassemia, hemoglobin E, and glucose-6-phosphate dehydrogenase deficiency in populations of malaria-endemic southern Shan State, Myanmar. Int J Hematol 2005; 82(2):119-123.
- ↑ Ali N, Anwar M, Ayyub M, Bhatti FA, Nadeem M, Nadeem A. Frequency of glucose-6-phosphate dehydrogenase deficiency in some ethnic groups of Pakistan. J Coll Physicians Surg Pak 2005; 15(3):137-141.
- ↑ Joseph R, Ho LY, Gomez JM, Rajdurai VS, Sivasankaran S, Yip YY. Mass newborn screening for glucose-6-phosphate dehydrogenase deficiency in Singapore. Southeast Asian J Trop Med Public Health 1999; 30 Suppl 2:70-71. PMID 11400790
- ↑ Rebholz CE, Michel AJ, Maselli DA, Saipphudin K, Wyss K. Frequency of malaria and glucose-6-phosphate dehydrogenase deficiency in Tajikistan. Malar J 2006; 5:51.
- ↑ Tanphaichitr VS. Glucose-6-phosphate dehydrogenase deficiency in Thailand; its significance in the newborn. Southeast Asian J Trop Med Public Health 1999; 30 Suppl 2:75-78.
- ↑ Tanphaichitr VS, Pung-Amritt P, Yodthong S, Soongswang J, Mahasandana C, Suvatte V. Glucose-6-phosphate dehydrogenase deficiency in the newborn: its prevalence and relation to neonatal jaundice. Southeast Asian J Trop Med Public Health 1995; 26 Suppl 1:137-141.
- ↑ Tanphaichitr VS, Mahasandana C, Suvatte V, Yodthong S, Pung-Amritt P, Seeloem J. Prevalence of hemoglobin E, alpha-thalassemia and glucose-6-phosphate dehydrogenase deficiency in 1,000 cord bloods studied in Bangkok. Southeast Asian J Trop Med Public Health 1995; 26 Suppl 1:271-274. PMID 8629122
- ↑ Verle P, Nhan DH, Tinh TT et al. Glucose-6-phosphate dehydrogenase deficiency in northern Vietnam. Trop Med Int Health 2000; 5(3):203-206.
- ↑ Moormann AM, Embury PE, Opondo J et al. Frequencies of sickle cell trait and glucose-6-phosphate dehydrogenase deficiency differ in highland and nearby lowland malaria-endemic areas of Kenya. Trans R Soc Trop Med Hyg 2003; 97(5):513-514.
- ↑ Bonafede RP, Botha MC, Beighton P. Glucose-6-phosphate dehydrogenase deficiency in the Greek population of Cape Town. S Afr Med J 1984; 65(14):547-549. PMID 6710260
- ↑ Kaddari F, Sawadogo M, Sancho J et al. [Neonatal screening of glucose-6-phosphate dehydrogenase deficiency in umbilical cord blood]. Ann Biol Clin (Paris) 2004; 62(4):446-450.
- ↑ Wolf BH, Schutgens RB, Nagelkerke NJ, Weening RS. Glucose-6-phosphate dehydrogenase deficiency in ethnic minorities in The Netherlands. Trop Geogr Med 1988; 40(4):322-330. PMID 3227552
- ↑ World Health Organization (WHO). Standardization of procedures for the study of glucose-6-phosphate dehydrogenase. WHO Tech. Rep. Ser. 1967, 366.
- ↑ Alving AS, Carson PE, Flanagan CL, Ickes CE. Enzymatic deficiency in primaquine-sensitive erythrocytes. Science 1956; 124(3220):484-485. PMID 13360274
- ↑ Persico MG, Toniolo D, Nobile C, D'Urso M, Luzzatto L. cDNA sequences of human glucose 6-phosphate dehydrogenase cloned in pBR322. Nature 1981; 294(5843):778-780.
Datenbanken
- Online Mendelian Inheritance in Man (OMIM) des National Center for Biotechnology Information (NCBI), National Library of Medicine (NLM), U.S.A. Datensatz +305900. Stand 21. Juli 2006. Informationen in Englisch.
- EntrezProtein des National Center for Biotechnology Information (NCBI), National Library of Medicine (NLM), U.S.A. Datensatz 120731. Stand 19. September 2006. Informationen in Englisch.
- SwissProt Datenbank, Universität Genf, Schweiz. Datensatz P11413. Stand 19. September 2006. Informationen in Englisch.
- Human Gene Mutation Database (HGMD), Institute of Medical Genetics, Cardiff, U.K. Datensatz G6PD. Stand 23. September 2006. Informationen in Englisch.

Bitte beachte den Hinweis zu Gesundheitsthemen! -
Wikimedia Foundation.