- Glykolyse
-
Übergeordnet Glucose-Stoffwechsel
EnergiestoffwechselGene Ontology AmiGO QuickGO Die Glykolyse (altgriechisch γλυκύς glykys ‚süß‘ und λύσις lysis ‚Auflösung‘) ist der schrittweise Abbau von Monosacchariden (Einfachzuckern) wie der D-Glucose (Traubenzucker), von der sich auch die Bezeichnung Glykolyse ableitet, bei Lebewesen. Sie ist der zentrale Prozess beim Abbau aller Kohlenhydrate in allen Eukaryoten, dazu gehören Tiere, Pflanzen und Pilze. Bei Bakterien und Archaeen ist Glykolyse ebenfalls verbreitet, manche Arten nutzen aber auch andere Stoffwechselwege um Glucose abzubauen, beispielsweise den Entner-Doudoroff-Weg (ED-Weg). Die Glykolyse ist ein zentraler Vorgang im Energiestoffwechsel und einer der wenigen Stoffwechselwege, den fast alle Organismen gemeinsam haben, was auf eine sehr frühe Entstehung hinweist.
Der Abbau erfolgt in zehn Einzelschritten. Dabei entstehen aus einem Glucosemolekül zwei Moleküle Pyruvat. Neben dem für das Freiwerden von Energie wichtigen Adenosintriphosphat (ATP) werden auch zwei Moleküle NADH erzeugt. Die Glykolyse wird nach ihren Entdeckern Gustav Embden, Otto Meyerhof und Jakub Karol Parnas auch Embden-Meyerhof-Parnas-Weg oder EMP-Weg genannt. Nicht mehr gebräuchlich ist die Bezeichnung FDP-Weg, die auf das Zwischenprodukt D-Fructose-1,6-bisphosphat (veraltet: Fructosediphosphat) zurückgeht.
Entdeckungsgeschichte
 Eduard Buchner erhielt 1907 den Nobelpreis für Chemie für seine Entdeckung der zellfreien Vergärung.
Eduard Buchner erhielt 1907 den Nobelpreis für Chemie für seine Entdeckung der zellfreien Vergärung.
Untersuchungen über den Abbau von Zucker gehen weit ins 19. Jahrhundert zurück und begannen ursprünglich mit der Erforschung der alkoholischen Gärung beziehungsweise später der Milchsäuregärung. Bei diesen Gärungen sind die Reaktionsschritte bis zur Bildung von Pyruvat identisch. 1837 wurde durch die Forscher Charles Cagniard-Latour[1], Theodor Schwann[2] und Friedrich Traugott Kützing[3] unabhängig voneinander nachgewiesen, dass der heutzutage als alkoholische Gärung bekannte Abbau von Glucose zu Ethanol durch Lebewesen, nämlich Hefen, verursacht wird.[4] Dass für den anaeroben Abbau von Zuckern die Stoffwechselprozesse lebender Hefezellen verantwortlich sind, war zum damaligen Zeitpunkt noch sehr umstritten. Vor allem die prominenten Chemiker Jöns Jakob Berzelius, Friedrich Wöhler und Justus von Liebig zählten zu den heftigsten Gegnern dieser Ansicht. Liebig postulierte beispielsweise, dass verwesendes Material „Schwingungen“ auf den zu vergärenden Zucker übertrage, welcher dadurch zu Ethanol und Kohlenstoffdioxid zerfalle.[5]
Dem Abbau von Zuckern in lebenden Hefezellen widmete sich ab 1857 auch der französische Forscher Louis Pasteur. 1860 veröffentlichte er eine Bestätigung der Ergebnisse von Cagniard-Latour, Kützing und Schwann und stellte sich damit gegen Liebigs Hypothese.[6] Außerdem beobachtete er, dass der Verbrauch an Glucose unter anaeroben Bedingungen höher ist als wenn den Hefen Sauerstoff zur Verfügung steht. Diese Beobachtung wird heutzutage als der „Pasteur-Effekt“ bezeichnet.
Zu jener Zeit herrschte die Lehrmeinung vor, dass nur eine den Lebewesen innewohnende „Lebenskraft“ (vis vitalis) die Umwandlung von Glucose zu Ethanol bewerkstelligen könne. 1858 schlug dagegen Moritz Traube vor, dass für den Abbau von Zuckern in Hefezellen allein chemische Prozesse, weniger die „Lebendigkeit“ als solche verantwortlich seien.[7] 1897 schließlich entdeckte Eduard Buchner, dass alkoholische Vergärung auch in einem zellfreien Hefeextrakt möglich ist.[8] Damit zeigte er, dass der Stoffwechselweg auch dann stattfinden kann, wenn die Zellen nicht mehr intakt sind. Man bezeichnet dies als in vitro. Das katalytisch wirksame Präparat bezeichnete er als „Zymase“, ohne zu wissen, dass mehrere Enzyme in den anaeroben Glucoseabbau involviert sind. Auch Marie von Mannasein zog im selben Jahr in einer Veröffentlichung ähnliche Schlüsse wie Buchner.[9] Jedoch konnte ihre Arbeit andere nicht überzeugen, da ihre Beweisführung unzureichend war.
 Arthur Harden leistete in Zusammenarbeit mit William John Young einen besonderen Beitrag in der Aufklärung der Glykolyse. Harden wurde zusammen mit von Euler-Chelpin 1929 der Nobelpreis für Chemie verliehen.
Arthur Harden leistete in Zusammenarbeit mit William John Young einen besonderen Beitrag in der Aufklärung der Glykolyse. Harden wurde zusammen mit von Euler-Chelpin 1929 der Nobelpreis für Chemie verliehen.Die Aufklärung der einzelnen Schritte der Glykolyse gelang ab Anfang des 20. Jahrhunderts. So konnten Arthur Harden und William John Young (1878–1942) Entscheidendes zur Aufklärung des glykolytischen Stoffwechselweges beitragen und veröffentlichten ihre Ergebnisse in einer Serie von Publikationen ab 1905. Unter anderem fanden sie heraus, dass isolierte Hefeextrakte Glucose nur langsam zu Ethanol und Kohlenstoffdioxid abbauten, wenn in den Extrakten kein anorganisches Phosphat vorhanden war.[10] Bei Zugabe von Phosphat jedoch konnte diese in vitro, also ohne lebende Zellen stattfindende Gärreaktion wieder schneller ablaufen.[11] Außerdem gelang es ihnen, Fructose-1,6-bisphosphat zu isolieren und nachzuweisen, dass es ein Zwischenprodukt der Glykolyse ist. Zudem trennten sie zellfreien Hefeextrakt mittels Dialyse in zwei Fraktionen auf.[12] Die Forscher bezeichneten die nicht dialysierbare Fraktion, das sind normalerweise größere Moleküle und Proteine, nach Buchner als „Zymase“. Sie war wärmeempfindlich. Die dialysierbare Fraktion besteht dagegen aus Ionen und kleinen Molekülen, die die Dialysemembran passieren können. Diese war wärmestabil und wurde „Cozymase“ genannt. Nur beide zusammen konnten eine Gärreaktion in vitro hervorrufen. Es stellte sich heraus, dass die Zymase ein Enzymgemisch war, während die Cozymase die für diese Enzyme nötigen Coenzyme enthielt.
1918 konnte Otto Meyerhof nachweisen, dass in der Milchsäuregärung in Muskeln die gleichen Coenzyme benötigt werden wie bei der alkoholischen Gärung.[13] Wegen der Kurzlebigkeit vieler Zwischenprodukte gestaltete sich die weitere Aufklärung des Stoffwechselweges als schwierig. Gustav Embden schlug 1932 eine erste biochemische Reaktionsfolge für die Glykolyse vor. Zwei Jahre später konnte Karl Lohmann im Labor Meyerhofs den Nachweis erbringen, dass der universelle Energieträger Adenosintriphosphat (ATP) bei der Glykolyse erzeugt wird. Meyerhofs Forschergruppe hatte Anteil an der Entdeckung etwa eines Drittels der an der Glykolyse beteiligten Enzyme.
Schließlich waren Ende der 1930er-Jahre durch die Arbeiten von Otto Warburg und Hans von Euler-Chelpin die Reaktionsschritte in Hefe aufgeklärt; Embden, Meyerhof und Jakub Karol Parnas arbeiteten dagegen mit Muskelzellen. Außerdem hatten auch Carl und Gerty Cori, Carl Neuberg, Robert Robinson sowie der unter Warburg tätige Erwin Negelein wesentlichen Anteil an der Aufklärung der Glykolyse.
Alle Schritte und Enzyme der Glykolyse sind seit den 1940er-Jahren bekannt. Genauere Untersuchungen der beteiligten Enzyme und ihrer Regulation folgten anschließend.
Bedeutung für die Zelle
Die Glykolyse ist der wichtigste Abbauweg der Kohlenhydrate im Stoffwechsel. Größtenteils werden alle Hexosen und Triosen durch diesen einen Stoffwechselweg metabolisiert und für den weiteren Abbau vorbereitet. Damit nimmt die Glykolyse einen zentralen Platz im katabolen Stoffwechsel ein. Die an den Reaktionen beteiligten Enzyme kommen in fast allen Lebewesen vor, so dass die Glykolyse auch universell ist. Die Glykolyse hat daneben auch noch weitere wichtige Funktionen:
Energiegewinnung unter anaeroben Bedingungen
In der Glykolyse wird Energie gewonnen und in Form von zwei Molekülen ATP je Molekül abgebauter D-Glucose bereitgestellt, unabhängig davon, ob Sauerstoff für die Atmungskette vorliegt oder nicht. Die Glykolyse erzeugt ungefähr ein Fünfzehntel so viel ATP auf ein Molekül D-Glucose wie der vollständige oxidative Abbau zu Kohlenstoffdioxid und Wasser im Citratzyklus und in der Atmungskette. Daher wird unter aeroben Bedingungen auch weniger Glucose verstoffwechselt, was bereits 1861 von Louis Pasteur bei Hefen beobachtet wurde (Pasteur-Effekt).
Da die Glykolyse auch unter anoxischen Bedingungen abläuft, eröffnet dies einige vorteilhafte Möglichkeiten im Stoffwechsel. Beispielsweise können Mikroorganismen in einem anoxischen Milieu auf diese Weise Energie gewinnen. Bei Wirbeltieren wird im Falle starker Muskelbeanspruchung manchmal mehr Sauerstoff verbraucht als in die Zellen transportiert wird. Daher muss die Zelle ihre Energie kurzfristig ausschließlich aus der Glykolyse beziehen. Dies ist häufig bei größeren Tieren wie Alligatoren, Krokodilen, Elefanten, Nashörnern, Walen und Robben der Fall, bei denen Sauerstoff für den oxidativen Abbau von Glucose nicht schnell genug bereitgestellt werden kann.[14] Auch beim Menschen wird Glucose in schnell kontrahierenden Muskelzellen im Zuge der Glykolyse und der Milchsäuregärung zu Lactat umgesetzt. Ein großer Vorteil der Glykolyse ist die Tatsache, dass ATP dabei 100 mal so schnell bereitgestellt werden kann wie über die oxidative Phosphorylierung in der Atmungskette.[15]
Pflanzen gewinnen ihre Energie entweder aus der Photosynthese oder aus der Atmungskette. Es gibt jedoch auch Situationen, in denen temporär Licht und Sauerstoff nicht zur Verfügung steht, beispielsweise bei der Imbibition während der Samenkeimung oder bei einer zeitweiligen Überflutung der Wurzeln mit Wasser. Unter diesen Bedingungen wird der lokale Stoffwechsel durch die Glykolyse aufrechterhalten.[16]
Glucose als einziger Brennstoff
 Erythrozyten decken ihren Energiebedarf ausschließlich aus der Glykolyse.
Erythrozyten decken ihren Energiebedarf ausschließlich aus der Glykolyse.Einige spezialisierte Zellen beziehen ihre Energie ausschließlich aus der Glykolyse. So sind beispielsweise Zellen im Gehirn und dem Nierenmark auf Glucose als Brennstoff angewiesen; Erythrozyten, denen die Mitochondrien und damit die Atmungskette fehlen, und Spermien[17] sowie schnell wachsende und sich teilende Tumorzellen gehören ebenfalls dazu. Otto Warburg entdeckte 1930, dass Tumorzellen eine sehr viel höhere Glykolyserate besitzen als gesunde Zellen. In der Positronen-Emissions-Tomographie wird dies genutzt, um Tumorgewebe bildlich darzustellen.
Bausteine für Zellmaterial
Die Glykolyse bereitet Glucose nicht nur für den oxidativen Abbau vor, sondern liefert auch Vorläufer für die Biosynthese anderer Verbindungen. So ist Pyruvat Ausgangsstoff für die Fettsäuresynthese und für manche Aminosäuren (L-Alanin, L-Valin und L-Leucin). Aus Dihydroxyacetonphosphat wird reduktiv Glycerin-3-phosphat gebildet, welches bei der Synthese von Lipiden eine Rolle spielt. Phosphoenolpyruvat ist Ausgangsstoff für die Biosynthese der aromatischen Aminosäuren L-Phenylalanin, L-Tryptophan und L-Tyrosin, während L-Serin aus 3-Phosphoglycerat gebildet wird.[18][19]
Bereitstellung von NADH
In der Glykolyse wird neben ATP auch das Reduktionsmittel NADH erzeugt. Dies wird entweder in der Atmungskette für einen weiteren ATP-Gewinn reoxidiert, oder als Reduktionsmittel für die Synthese anderer Moleküle verwendet – zumindest zum Zwecke der NAD+-Regeneration in Gärungen.
Reaktionsschritte
Zelluläre Lokalisation
Die Glykolyse findet im Zytoplasma einer Zelle statt. In multizellulären Organismen – wie beispielsweise dem Menschen – wird die Glykolyse in allen (differenzierten) Zelltypen durchgeführt. Pflanzen betreiben die Glykolyse auch zusätzlich in den Plastiden.[20]
Allgemeiner Überblick
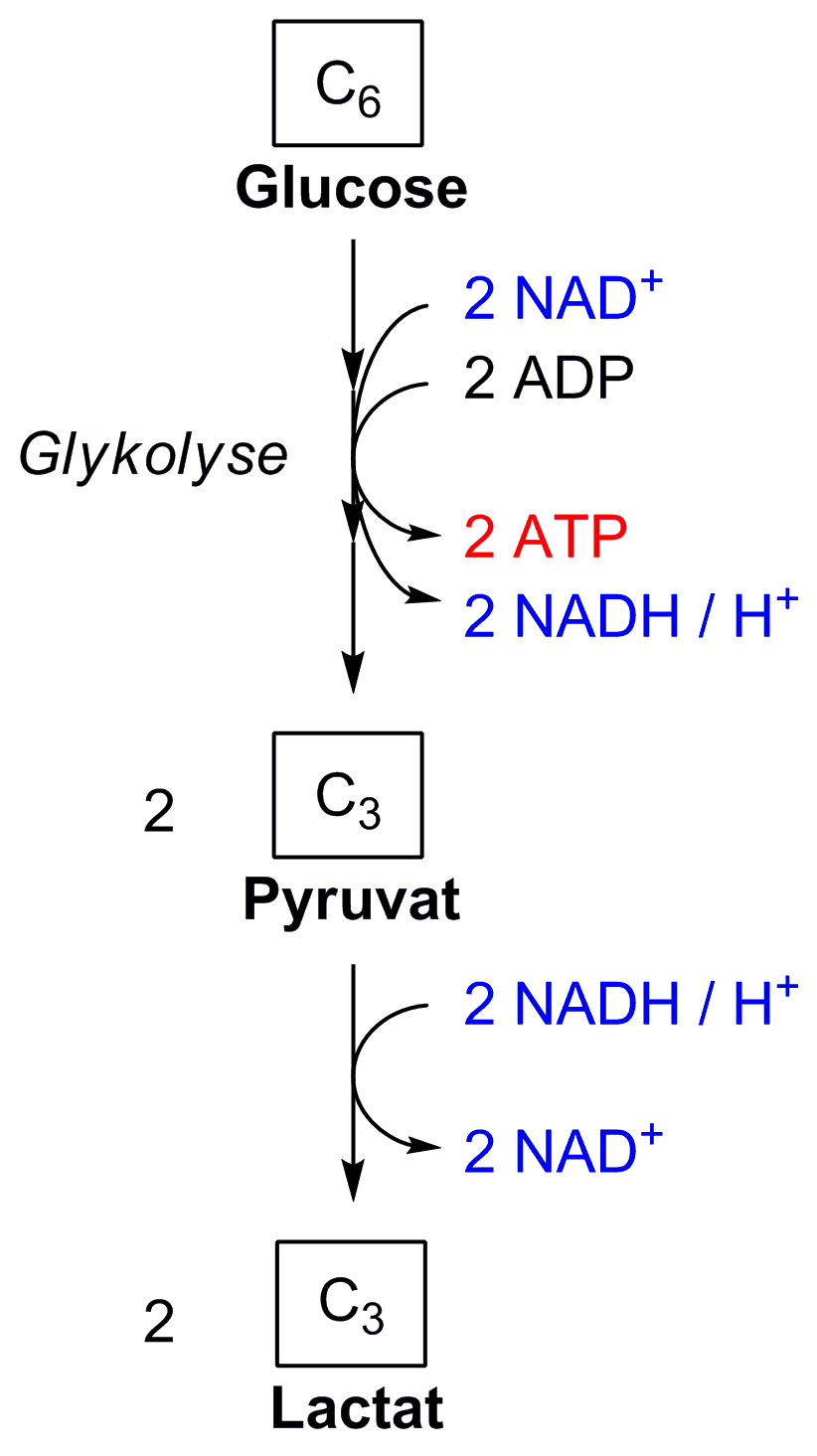
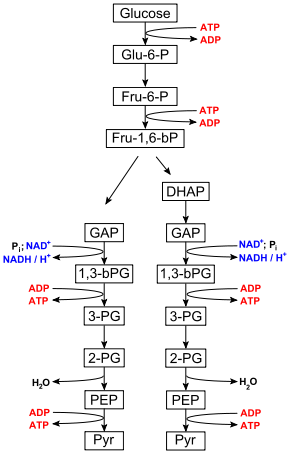 Ablauf der Glykolyse: Ein Molekül Glucose wird zu zwei Molekülen Pyruvat umgesetzt, dabei werden zunächst zwei Moleküle ATP investiert. Im späteren Verlauf der Glykolyse werden vier Moleküle ATP und zwei Moleküle NADH erzeugt. Einzelheiten bitte dem Text entnehmen. Abkürzungen:
Ablauf der Glykolyse: Ein Molekül Glucose wird zu zwei Molekülen Pyruvat umgesetzt, dabei werden zunächst zwei Moleküle ATP investiert. Im späteren Verlauf der Glykolyse werden vier Moleküle ATP und zwei Moleküle NADH erzeugt. Einzelheiten bitte dem Text entnehmen. Abkürzungen:
Glu-6-P = Glucose-6-phosphat
Fru-6-P = Fructose-6-phosphat
Fru-1,6-bP = Fructose-1,6-bisphosphat
DHAP = Dihydroxyacetonphosphat
GAP = Glycerinaldehyd-3-phosphat
1,3-bPG = 1,3-Bisphosphoglycerat
3-PG = 3-Phosphoglycerat
2-PG = 2-Phosphoglycerat
PEP = Phosphoenolpyruvat
Pyr = PyruvatDer Abbau von Glucose bis zu Pyruvat läuft sowohl unter Sauerstoffmangelbedingungen (anaerob) als auch bei ausreichendem Sauerstoffangebot (aerob) gleichartig ab. Im Gegensatz zur Atmungskette wird kein Sauerstoff (O2) verbraucht.
Die Glykolyse lässt sich in zwei Phasen unterteilen. Die erste Phase ist eine Vorbereitungsphase, bei der zunächst Energie in Form von ATP investiert wird. Sie besteht aus der Spaltung der Hexose D-Glucose in zwei Triosephosphate: Dihydroxyacetonphosphat (DHAP) und Glycerinaldehyd-3-phosphat (GAP) (vgl. Abbildung). Hierbei wird DHAP in GAP für die zweite Phase isomerisiert. Dadurch wird der Zucker für den eigentlichen Abbau vorbereitet.
In der zweiten Phase werden zwei Moleküle GAP über mehrere Zwischenschritte in zwei Moleküle Pyruvat (Pyr) umgesetzt. Dabei werden zwei Moleküle NADH sowie vier Moleküle ATP gebildet. Diese Phase liefert somit Energie und 4 Reduktionsäquivalente in Form von NADH.
Die Gesamtbilanz der Glykolyse kann damit wie folgt formuliert werden:
Vorbereitungsphase
Der erste Schritt der Glykolyse ist die Phosphorylierung von D-Glucose (Glc) zu Glucose-6-phosphat (G6P). In Abhängigkeit vom Zelltyp wird diese Reaktion durch eine Hexokinase oder Glucokinase (= Hexokinase IV) katalysiert, bei der ein Molekül ATP investiert wird. Dies hat zwei Vorteile: Zum einen ist die Zellmembran zwar durchlässig für Glucose, nicht aber für das durch die Phosphorylierung entstehende Glucose-6-phosphat. Dadurch reichert es sich in der Zelle an. Zum anderen verringert sich durch Phosphorylierung der Glucose die Glucosekonzentration in der Zelle, während die Konzentration an G6P umgekehrt ansteigt. Die initiale Phosphorylierung bewirkt damit, dass innerhalb der Zelle weniger Glucose vorliegt als außerhalb der Zelle. Da die intrazelluläre Glucosekonzentration im Gleichgewicht zu der extrazellulären steht, strömt weitere Glucose durch dieses entstandene Konzentrationsgefälle in die Zelle ein. Infolgedessen ist die Aufnahme von Glucose begünstigt.
In Bakterien wird die Phosphorylierung im ersten Schritt der Glykolyse nicht durch Hexo- beziehungsweise Glucokinasen, sondern durch das Phosphoenolpyruvat (PEP)-abhängige Zucker-Phosphotransferasesystem katalysiert.[21]
Glucose-6-phosphat wird dann von der Glucose-6-phosphat-Isomerase in das isomere Fructose-6-phosphat (F6P) umgebaut. Das Enzym bevorzugt die Bindung des alpha-Anomers der G6P, als Reaktionsprodukt entsteht α-D-Fructose-6-phosphat. Unter Standardbedingungen liegt zwar das Gleichgewicht der Isomerisierungsreaktion auf Seite des G6P. Aber da F6P schnell weiterreagiert, wird dieses dem Reaktionssystem entzogen, so dass sich kein Gleichgewicht einstellt und die Isomerisierungsreaktion zu Gunsten des F6P abläuft.
ATP ADP

Hexokinase
oder
Glucokinase
Glucose-
6-phosphat-
Isomerase

α-D-Glucose α-D-Glucose-6-phosphat α-D-Fructose-6-phosphat Fructose-6-phosphat wird danach unter Einwirkung des ersten Schlüsselenzyms der Glykolyse, Phosphofructokinase 1, mit einem Molekül ATP zu Fructose-1,6-bisphosphat (F1,6bP) phosphoryliert, wobei ADP entsteht. Das Enzym bevorzugt das beta-Anomer der F6P, in der Vorreaktion ist dagegen das alpha-Anomer entstanden. Dies stellt jedoch kein Problem dar, da die beiden Anomere im Gleichgewicht stehen. In anaeroben Bakterien, manchen Pflanzen, primitiven Eukaryoten und manchen Archaeen wird dieser Schritt von einer Pyrophosphat-abhängigen Phosphofructokinase (EC 2.7.1.90) katalysiert, bei der Pyrophosphat (PPi) anstatt ATP verwendet wird.[22]
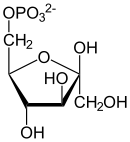
ATP ADP
Phospho-
fructo-
kinase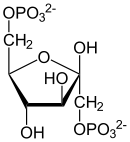
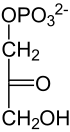
β-D-Fructose-6-phosphat β-D-Fructose-1,6-bisphosphat Die damit erneute Investition von Energie ist aus zwei Gründen günstig und notwendig: Zum einen macht dieser Schritt – neben der Glucokinase sowie der Pyruvatkinase – die Glykolyse unter physiologischen Bedingungen unumkehrbar. Zum anderen ermöglicht die Spaltung der Hexose die Bildung von zwei phosphorylierten Triosen für den weiteren Abbau, Dihydroxyacetonphosphat (DHAP) und Glycerinaldehyd-3-phosphat (GAP). Die Kohlenstoffatome C1-C3 der F1,6bP finden sich in DHAP, während die C-Atome in GAP aus der C4-C6 Einheit der F1,6bP stammen.
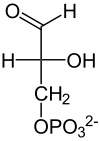
Die Spaltungsreaktion ist unter Standardbedingungen sehr ungünstig (ΔG0’ = +24 kJ/mol) und würde nicht ablaufen. Durch die schnelle Metabolisierung beider Reaktionsprodukte läuft sie aber unter physiologischen Bedingungen im annähernden Gleichgewicht ab. Dihydroxyacetonphosphat wird noch von der Triosephosphatisomerase (TIM) in D-Glycerinaldehyd-3-phosphat umgewandelt. Diese stereospezifische Isomerisierung in Richtung GAP wird dadurch begünstigt, dass GAP in der Glykolyse weiter abgebaut wird und damit die Konzentration in der Zelle niedrig gehalten wird. Ohne weitere Metabolisierung würde das Gleichgewicht zwischen DHAP und GAP stark auf Seiten des Ketons liegen (22:1).[23]
Aldolase
Triose-
phosphat-
Isomerase
β-D-Fructose-1,6-bisphosphat Dihydroxy-
aceton-
phosphatD-Glycerin-
aldehyd-
3-phosphatAmortisierungsphase
Jedes der beiden resultierenden Glycerinaldehyd-3-phosphat-Moleküle wird zu Beginn der Amortisierungsphase der Glykolyse durch eine Glycerinaldehyd-3-phosphat-Dehydrogenase (GAPDH) oxidiert. Bei der Reaktion wird NAD+ zu NADH reduziert. Die Oxidation der Aldehydgruppe (GAP) zur Carboxygruppe ist energetisch sehr günstig. Sie wird ausgenutzt, um anorganisches Phosphat mit der Carboxygruppe zu verknüpfen. Es entsteht dadurch das gemischte Säureanhydrid 1,3-Bisphosphoglycerat (1,3-BPG). Das Gleichgewicht dieser Reaktion ist zwar auf Seiten des Eduktes GAP gegenüber 1,3-BPG (10:1).[23] Durch die schnelle Umsetzung des Produktes wird aber die Einstellung des Gleichgewichts verhindert und ständig 1,3-BPG gebildet, außerdem begünstigt eine hohe Konzentration an NAD+ gegenüber NADH die Umsetzung in eine Richtung.
Ein in Erythrozyten vorhandener alternativer Nebenweg, vom 1,3-Bisphosphoglycerat zum 3-Phosphoglycerat, ist der über das Intermediat 2,3-Bisphosphoglycerat verlaufende Rapoport-Luebering-Zyklus, dessen zentrales Enzym die trifunktionale Bisphosphoglyceratmutase ist.
NAD+ NADH
+ Pi + H+

Glycerinaldehyd-
3-phosphat-
Dehydrogenase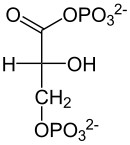
D-Glycerinaldehyd-3-phosphat D-1,3-Bisphosphoglycerat Im nächsten Schritt erzeugt die Phosphoglyceratkinase je ein Molekül ATP bei der Umwandlung von 1,3-Bisphosphoglycerat zu 3-Phosphoglycerat durch Übertragung eines Phosphatrests auf ADP. Die bei der vorangegangenen Oxidation frei gewordene Energie wird also konserviert, indem ATP aufgebaut wird. Die hier stattfindende Bildung von ATP aus ADP ist ein Beispiel der Substratphosphorylierung. Falls die Zelle bereits viel ATP (und damit wenig ADP) hat, hält die Reaktion an dieser Stelle an, bis wieder genügend ADP zur Verfügung steht. Diese Feedbackregulation ist wichtig, da ATP relativ schnell zerfällt, wenn es nicht genutzt wird. Überproduktion von ATP wird somit verhindert.
Die Energiebilanz der Glykolyse ist an diesem Schritt ausgeglichen: zwei Moleküle ATP wurden verbraucht und zwei wieder gewonnen
Phospho-
glycerat-
kinase
ADP ATP
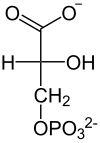
D-1,3-Bisphosphoglycerat D-3-Phosphoglycerat  Die Enolform des Pyruvates
Die Enolform des PyruvatesEine Kofaktor-unabhängige Phosphoglyceratmutase (PGM) katalysiert dann die Umwandlung von 3-Phosphoglycerat zu 2-Phosphoglycerat. Bei dem Vorgang wird die Phosphatgruppe zwischenzeitlich auf einen Aminosäurerest des Enzyms übertragen. In Erythrozyten wird diese Reaktion von einer Cofaktor-abhängigen PGM katalysiert, bei der 2,3-Bisphosphogylcerat als Zwischenprodukt gebildet wird.
2-Phosphoglycerat wird dann mit Hilfe der Enolase zu Phosphoenolpyruvat (PEP) dehydratisiert. Daher nennt man das Enzym auch 2-Phosphoglycerat-Dehydratase. PEP ist eine phosphorylierte Verbindung mit einem sehr hohen Gruppenübertragungspotential. Dies wird im letzten Schritt der Glykolyse genutzt, um ein weiteres Molekül ATP zu gewinnen. Hierbei katalysiert eine Pyruvatkinase (PK) unter ATP-Gewinn die Umsetzung von PEP zu Pyruvat (= Anion der Brenztraubensäure). Dabei entsteht jedoch nicht Pyruvat direkt, sondern das im Gleichgewicht stehende Enolpyruvat.[24] Bei pH 7 liegt das Gleichgewicht auf Seiten der Ketonform. Auch dieser Schritt ist ADP-reguliert, es ist die dritte, irreversible Reaktion im Verlauf der Glykolyse.
Phospho-
glycerat-
Mutase
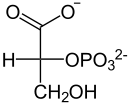
−H2O
Enolase
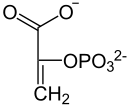
ADP ATP
Pyruvatkinase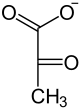
D-3-Phosphoglycerat D-2-Phosphoglycerat Phosphoenolpyruvat Pyruvat Bei Phosphatmangel können Pflanzen PEP ohne ATP-Gewinn zu Pyruvat hydrolysieren, was in den Vakuolen stattfindet. Das beteiligte Enzym ist eine PEP-Phosphatase (EC 3.1.3.60), die anorganisches Phosphat freisetzt und damit dem Phosphatmangel entgegensteuert.[25]
Regeneration des Cofaktors NADH
In der Glykolyse werden pro Durchgang zwei Moleküle NAD+ zu NADH reduziert. Meistens ist die zelluläre Konzentration an NAD+ sehr niedrig, so dass es ohne eine Reoxidation schnell verbraucht wäre. Infolgedessen muss NAD+ wieder regeneriert werden, da sonst die Glykolyse zum Erliegen kommt. Wie das geschieht, hängt davon ab, ob anoxische oder oxische Bedingungen vorliegen. Zudem beeinflusst dies den weiteren Abbauweg des Pyruvates.
Oxische Bedingungen
Unter oxischen Bedingungen wird Pyruvat im Pyruvatdehydrogenase-Komplex zu Acetyl-CoA oxidativ decarboxyliert. Hierbei entstehen ein Molekül Kohlenstoffdioxid und ein Molekül NADH. Acetyl-CoA tritt anschließend im Citratzyklus ein, in der es vollständig zu zwei Molekülen Kohlenstoffdioxid oxidiert wird. Bei diesen Oxidationsschritten entstehen weitere Reduktionsäquivalente. Diese und die aus der Glykolyse stammenden werden schließlich im Zuge der Atmungskette unter Verbrauch von Sauerstoff reoxidiert und stehen somit wieder der Glykolyse und dem Citratzyklus zur Verfügung. Gleichzeitig werden bei diesen Schritten weitere Moleküle ATP gebildet. Während Prokaryoten etwa insgesamt 38 Moleküle ATP je Mol Glucose erzeugen können, hängt die Bilanz bei Eukaryoten vom Weg ab, auf dem im Cytosol gebildetes NADH die Mitochondrienmembran passiert (Malat-Aspartat-Shuttle beziehungsweise Glycerin-3-phosphat-Shuttle).
In Eukaryoten findet der Citratzyklus in der Matrix des Mitochondriums, die Glykolyse hingegen im Cytosol statt. NAD+ und NADH können nicht frei durch die innere Membran des Mitochondriums diffundieren, außerdem fehlen spezielle Translokatoren. Der Austausch der Reduktionsäquivalente findet daher entweder durch den Malat-Aspartat-Shuttle oder den Glycerin-3-phosphat-Shuttle statt.
In der Literatur werden manchmal die Glykolyse und der Folgeabbau von Pyruvat zu Kohlenstoffdioxid durch die Prozesse des Citratzyklus und der Atmungskette fälschlicherweise als aerobe Glykolyse zusammengefasst. Die Glykolyse endet jedoch mit der Entstehung von Pyruvat und findet sowohl unter oxischen als auch anoxischen Bedingungen statt.
Anoxische Bedingungen
→ siehe auch Hauptartikel alkoholische Gärung, Milchsäuregärung
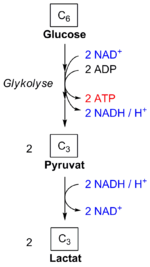 Bei der homofermentativen Milchsäuregärung wird das in der Glykolyse verbrauchte NAD+ in einer Folgereaktion wieder regeneriert.
Bei der homofermentativen Milchsäuregärung wird das in der Glykolyse verbrauchte NAD+ in einer Folgereaktion wieder regeneriert.Steht Sauerstoff nicht oder nur begrenzt zur Verfügung, kann Pyruvat reduktiv weiter umgesetzt werden, beispielsweise in der Milchsäuregärung oder in der alkoholischen Gärung. In der Milchsäuregärung wird Pyruvat zu L-Lactat reduziert, in der alkoholischen Gärung wird es zu Ethanol decarboxyliert und reduziert. In beiden Fällen wird NADH zu NAD+ oxidiert und liegt für weitere Runden der Glykolyse bereit. In diesen Gärungsschritten wird aber im Gegensatz zum aeroben Abbauweg kein ATP gebildet.
In der alkoholischen Gärung bilden Hefen in zwei Reaktionen aus Pyruvat Ethanol, welches durch zwei Enzyme, die Pyruvatdecarboxylase (EC 4.1.1.1) und die Alkoholdehydrogenase, katalysiert wird. Dabei wird das durch die Glykolyse angefallene NADH zu NAD+ reoxidiert. Andere Bakterien wie beispielsweise Milchsäurebakterien oder auch Muskelzellen im Menschen betreiben die Milchsäuregärung (vgl. auch Abbildung rechts). Hierbei wird Pyruvat durch eine Lactatdehydrogenase mittels NADH + H+ zu Lactat und NAD+ reduziert, so dass die Glykolyse wieder ablaufen kann. Diese Reaktion ist sowohl unter Standardbedinungen als auch unter physiologischen Bedingungen stark exergonisch (ΔG0′ = –25 kJ/mol beziehungsweise ΔG = –14,8 kJ/mol)[26]
Die (homofermentative) Milchsäuregärung wird teilweise als anaerobe Glykolyse bezeichnet. Dies ist jedoch irreführend, da die Glykolyse mit dem Entstehen von Pyruvat endet und unter sowohl oxischen als auch anoxischen Bedingungen stattfindet.
Energetische Aspekte
Gleichgewichtslage
Schritt Reaktion in der Glykolyse ΔG0’ [kJ/mol][27][26] ΔG [kJ/mol][26] 1 Glucose + ATP → Glucose-6-P + ADP −16,7 −33,9 2 Glucose-6-P ⇌ Fructose-6-P +1,7 −2,9 3 Fructose-6-P + ATP → Fructose-1,6-bP + ADP −14,2 −18,8 4 Fructose-1,6-bP ⇌ DHAP + G-3-P +23,9 −0,2 5 DHAP ⇌ GAP +7,6 +2,4 6 GAP + Pi + NAD+ ⇌ 1,3-Bis-P-glycerat + NADH + H+ +6,3 −1,3 7 1,3-Bis-P-glycerat + ADP ⇌ 3-P-glycerat + ATP −18,9 +0,1 8 3-Phosphoglycerat ⇌ 2-Phosphoglycerat +4,4 +0,8 9 2-P-glycerat ⇌ PEP + H2O +7,5[27] beziehungsweise +1,8[26] +1,1 10 PEP + ADP → Pyruvat + ATP −31,7 −23,0 Die meisten Reaktionen der Glykolyse sind unter Standardbedingungen bei einem pH-Wert von 7 energetisch ungünstig. So ist häufig die Änderung der freien Enthalpie G0’ positiv, so dass jene Reaktionen endergon sind und nicht ablaufen würden (vgl. Tabelle ΔG0’-Werte). Die Glykolyse würde bereits im vierten Schritt enden.
Metabolit Konzentration [mM][28] Glucose 5,0 Glucose-6-P 0,083 Fructose-6-P 0,014 Fructose-1,6-bP 0,031 DHAP 0,140 GAP 0,019 1,3-Bis-P-glycerat 0,001 3-PG 0,120 2-PG 0,030 PEP 0,023 Pyruvat 0,051 Pi 0,001 Definitionsgemäß entspricht die Stoffmengenkonzentration der Reaktanten unter solchen Bedingungen jeweils 1 mol·l−1. Dies kann aber nicht als Grundlage einer Berechnung dienen, da lebende Zellen solch hohe Konzentrationen nicht erzeugen beziehungsweise aufrechterhalten können. Für eine aussagekräftige Beurteilung müsste man dagegen die tatsächlichen Stoffmengenkonzentrationen kennen. Misst man diese unter physiologischen Bedingungen, kann die Änderung der freien Enthalpie G neu berechnet werden (vgl. Tabelle ΔG-Werte, Metabolitkonzentration).
Für die Berechnung dieser Werte bieten sich insbesondere Erythrozyten an.[29] Erythrozyten beziehen ihre gesamte Energie ausschließlich aus der Glykolyse. Dabei finden auch alle übrigen zellulären Reaktionen im Cytoplasma statt, da sie weder über Mitochondrien, einen Zellkern noch ein endoplasmatisches Retikulum verfügen. Dies erleichtert außerdem die Trennung der Zellbestandteile. Ohne Mitochondrien finden auch keine Reaktionen der Atmungskette und des Citratzyklus statt. Eine Quantifizierung der daran beteiligten Coenzyme wäre sonst erheblich erschwert. Schließlich nimmt der Pentosephosphatweg nur einen geringen Anteil am Stoffwechsel von Erythrozyten ein, es erfolgt auch keine Protein- und keine Lipidbiosynthese. Damit können die glykolytischen Zwischenprodukte leicht isoliert und bestimmt werden.
1965 wurden die Stoffmengenkonzentrationen (steady state) glykolytischer Intermediate aus menschlichen Erythrozyten bestimmt (vgl. Tabelle rechts).[30] Es ergab sich, dass gewisse Intermediate in sehr niedrigen Konzentrationen vorliegen.[31] Unter Berücksichtigung dieser Konzentrationen ändert sich die Gleichgewichtslage der korrespondierenden Reaktionen derart, dass unter physiologischen Bedingungen die gesamte Glykolyse bis auf drei Reaktionen reversibel verläuft (ΔG ungefähr 0 kJ·mol−1).
Bei jenen Reaktionen bleibt die Stoffmengenkonzentration deswegen so niedrig, weil die erzeugten Produkte schnell umgesetzt und abschließend durch irreverbibe Reaktionen dem System entzogen werden. Diese drei irreversiblen Reaktionen werden von den Schlüsselenzymen Glucokinase beziehungsweise Hexokinase, Phosphofructokinase 1 sowie Pyruvatkinase katalysiert. Durch die schnelle, irreversible Umsetzung mittels eines der Schlüsselenzyme werden die Stoffmengenkonzentrationen der vorher erzeugten Produkte ausreichend abgesenkt – die Glykolyse kann in eine Richtung ablaufen.
Bei der Berechnung der Gleichgewichtslage unter physiologischen Bedingungen ergeben sich für manche Reaktionen kleine, aber dennoch positive ΔG-Werte, beispielsweise bei der Isomerisieruug von 3-Phosphoglycerat zu 2-Phosphoglycerat (ΔG = +1,1 kJ·mol−1). Streng genommen können diese Werte nicht ganz stimmen, da die Vorwärtsreaktion nur bei negativen ΔG-Werten stattfinden kann. Da die Glykolyse aber stattfindet, nimmt man an, dass für diesen Widerspruch Messfehler bei der Bestimmung der Stoffmengenkonzentrationen verantwortlich sind.[32]
Aus dem Vorhandensein dreier Kontrollpunkte ergeben sich zwei Konsequenzen. Erstens kann die Glykolyse an jenen Stellen effektiv reguliert werden, so dass sie abhängig vom Energiestatus der Zelle schnell an- beziehungsweise abgeschaltet werden kann. Zweitens ermöglicht die vorliegende Gleichgewichtslage auch die Umkehrreaktion der Glykolyse, die Gluconeogenese. Bis auf drei Enzyme werden hierbei alle Enzyme der Glykolyse verwendet.
Effizienz
Unter Standardbedingungen wird bei der Umsetzung von D-Glucose zu zwei Molekülen Lactat 183,6 kJ/mol Energie frei (ΔG0’ = −183,6 kJ/mol):
Für den Aufbau von zwei Molekülen ATP aus jeweils zwei Molekülen ADP sowie anorganischem Phosphat (Pi) werden 61,0 kJ/mol benötigt:
Da die Glykolyse diese zwei Reaktionen durch Substratkettenphosphorylierung koppelt, wird somit eine Energie von 122,6 kJ·mol/mol frei:
ΔG0’ = (−183,6 + 61) kJ·mol−1 = −122,6 kJ·mol−1
Unter Standardbedingungen werden beim anaeroben Abbau von Glucose zu Lactat damit 33 % der verfügbaren Energie genutzt, um zwei Moleküle ATP aufzubauen. Da unter physiologischen Bedingungen etwa 50 kJ·mol−1 für den Aufbau von ATP benötigt werden, ist die Energieausbeute auch etwas höher, etwa 50 %.[33]
Regulation
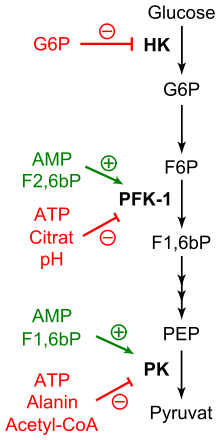 Die Regulation der Glykolyse in der Übersicht. Effektoren, die die Hexokinase (HK), Phosphofructokinase-1 (PFK-1) beziehungsweise die Pyruvatkinase (PK) aktivieren, sind in grün hervorgehoben. Metabolite, die diese Enzyme inhibieren, sind in rot dargestellt. Bitte auch Text beachten.
Die Regulation der Glykolyse in der Übersicht. Effektoren, die die Hexokinase (HK), Phosphofructokinase-1 (PFK-1) beziehungsweise die Pyruvatkinase (PK) aktivieren, sind in grün hervorgehoben. Metabolite, die diese Enzyme inhibieren, sind in rot dargestellt. Bitte auch Text beachten.Die Glykolyse dient der Bereitstellung von Energie, insbesondere wenn das entstehende Pyruvat unter aeroben Bedingungen weiter abgebaut wird. Liegt dagegen ein energetisch günstiger Zustand vor, wird Glucose gespeichert und im Zuge des Anabolismus unter Energieverbrauch in andere Metabolite umgewandelt.
Die Regulation der Glykolyse ist daher von entscheidender Bedeutung. Sie sollte beispielsweise nicht parallel zu deren Umkehrreaktion, der Gluconeogenese, ablaufen. In so einem Falle spricht man von einem „Leerlaufprozess“, der sinnlos ATP verbraucht und damit unproduktiv ist. Als Ausnahme sei die Wärmeerzeugung in Hummeln erwähnenswert, die durch beabsichtigte, gegenläufige Phosphorylierung und Dephosphorylierung von Fructose-6-phosphat zu Fructose-1,6-bisphosphat und umgekehrt Wärme erzeugen.[34]
Biochemisch betrachtet werden irreversible Reaktionen kontrolliert. Unter physiologischen Bedingungen gibt es drei Reaktionen in der Glykolyse, die irreversibel sind. Sie werden von der Hexo- beziehungsweise Glucokinase, von der Phosphofructokinase-1 und der Pyruvatkinase katalysiert und sind damit Ziel einer Regulation.
Hexo- und Glucokinase
Die Hexokinase ist das erste Enzym in der Glykolyse, dessen Aktivität reguliert wird. Es phosphoryliert unter ATP-Verbrauch Glucose zu Glucose-6-phosphat (G6P), kann aber auch andere Hexosen als Substrat verwenden. G6P ist das Endprodukt der Hexokinasereaktion und inhibiert als solches das Enzym allosterisch.[35]
Das in der Leber vorkommende Isoenzym der Hexokinase, die Glucokinase, wird nicht durch das Produkt G6P inhibiert. Im Gegensatz zur Hexokinase zeigt es auch einen höheren KM-Wert. Daher löst die Glucokinase erst bei sehr hohen Glucosekonzentrationen die Hexokinase ab. Unter diesen Bedingungen wird G6P durch nachfolgende Schritte aber als Glykogen gespeichert und nicht in der Glykolyse abgebaut, denn G6P wird auch für andere Stoffwechselwege abgezweigt. Die Leber fungiert daher als Homöostat des Blutzuckerspiegels, da sie diesen durch den Auf- beziehungsweise Abbau von Glucose aufrechterhält.[36]
Ein leberspezifisches, regulatorisches Protein kann reversibel an die Glucokinase binden und sie dadurch hemmen. Das Binden dieses Regulators an die Glucokinase findet im Zellkern statt, so dass die gehemmte Glucokinase dort inaktiv verbleibt und nicht durch andere cytosolische Effektoren beeinflusst werden kann. Diese Bindung wird dann verstärkt, falls das Enzym allosterisch durch Fructose-6-phosphat modifiziert wurde. Dagegen bewirkt Glucose ein Ablösen dieses Leberproteins. Bei einer hohen Blutzuckerkonzentration dominiert Glucose, so dass die Dissoziation des Regulators ermöglicht und Glucose zu Glucose-6-phosphat phosphoryliert wird. Sinkt der Blutzuckerspiegel aber zu sehr ab (unter 5 mmol·l−1), erfolgt die Hemmung der Glucokinase – vermittelt durch Fructose-6-phosphat. Glucose wird nicht mehr phosphoryliert und kann die Leber wieder verlassen, um anderen Organen zur Verfügung zu stehen.[36]
Schließlich wird die Glucokinase auf Ebene der Transkription reguliert.[35] Hierbei beeinflusst das Hormon Insulin die Menge an Glucokinase in der Leber. Eine Stoffwechselstörung liegt bei Patienten mit Diabetes mellitus vor, da sie Insulin nicht ausreichend herstellen können. Bei ihnen ist die Menge an Glucokinase zu niedrig, sie tolerieren nicht eine hohe Blutzuckerkonzentration und weisen nur wenig Glucokinase in der Leber auf.
Phosphofructokinase-1
Der wichtigste Kontrollpunkt der Glykolyse ist die Phosphorylierung von Frc-6-P zu Frc1,6-bP durch die Phosphofructokinase-1 (PFK-1). Er stellt den ersten wirklichen glykolysespezifischen Schritt dar und ist unter physiologischen Bedingungen irreversibel.[37] Das Enzym weist zwei Bindungsstellen für ATP auf. Neben einer hochaffinen Substratbindestelle verfügt die PFK-1 auch über eine regulatorische Seite. So kann ATP sowohl als Substrat dienen, als auch die PFK-1 allosterisch hemmen. Bei ausreichend hohen ATP-Konzentrationen wird der KM-Wert des Enzyms erhöht. Dies senkt die Aktivität der PFK-1, so dass die Glykolyse gedrosselt wird. Dennoch schwanken die ATP-Konzentrationen einer Zelle nur geringfügig, so dass ATP alleine nicht ausreichend wäre für eine genaue Regulation. Daher hängt die Aktivität der PFK-1 auch von der AMP-Konzentration ab und spiegelt die energetische Versorgung der Zelle wider. Falls diese ausreichend hoch ist, wird das Enzym gehemmt, andernfalls wird die Aktivität der PFK-1 erhöht, um mehr ATP zu bilden.
Auch Citrat inhibiert die PFK-1 allosterisch. Citrat ist ein Schlüsselmetabolit des Citratzyklus, dessen primärer Zweck die Erzeugung von Energie unter aeroben Bedingungen ist. Alternativ können aus dem Citratzyklus verschiedene Vorläufermoleküle entnommen werden. Falls viel Citrat vorliegt, ist der Citratzyklus gesättigt. Daher inhibiert Citrat die PFK-1 im Sinne einer Endprodukthemmung, so dass die Glykolyse den Citratzyklus weniger stark speist.
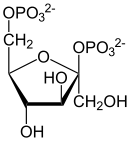 β-D-Fructose-2,6-bisphosphat, ein potenter Aktivator der Phosphofructokinase-1
β-D-Fructose-2,6-bisphosphat, ein potenter Aktivator der Phosphofructokinase-1Die Aktivität der Phosphofructokinase-1 wird auch durch den pH-Wert beeinflusst. Ein niedriger pH-Wert hemmt das Enzym und drosselt die Glykolyse. Dies passiert beispielsweise bei starker Muskelbeanspruchung, bei der viel Lactat entsteht. Dieses senkt den pH-Wert in den Zellen.[38]
Schließlich wird PFK-1 in mikromolaren Konzentrationen durch β-D-Fructose-2,6-bisphosphat (F-2,6-bP) allosterisch aktiviert.[38] Durch F-2,6-bP wird damit die Glykolyse gefördert, während sie die Fructose-1,6-bisphosphatase inhibiert. Dies ist das Enzym, das an dieser Stelle die Rückreaktion in der Gluconeogenese katalysiert. Unter physiologischen Bedingungen verbleibt das Enzym ohne F-2,6-bP in einem praktisch inaktiven Zustand.[36] Nach Binden von F-2,6-bP an PFK-1 wird auch die Affinität der beiden Inhibitoren ATP und Citrat reduziert.
In Bakterien kommt Fructose-2,6-bisphosphat als Aktivator der PFK-1 nicht vor.[38]
Pyruvatkinase
Der letzte Schritt in der Glykolyse ist irreversibel und wird von der Pyruvatkinase (PK) katalysiert. Diese wird zwar auch reguliert, aber im Gegensatz zu den anderen beiden Enzymen in vergleichsweise untergeordneter Weise. Fructose-1,6-bisphosphat und AMP stimulieren die PK, während ATP, Acetyl-CoA und L-Alanin diese allosterisch hemmen. Das in der Leber und Darm vorherrschende Isoenzym (L-Form) kann im Gegensatz zu der im Muskel vorkommenden M-Form zusätzlich durch die Proteinkinase A phosphoryliert werden. Die Aktivität der Proteinkinase A wird hormonell durch Glukagon vermittelt. In phosphorylierter Form wird dieses Isoenzym dann vergleichsweise stärker durch ATP und Alanin inhibiert als die unmodifizierte PK.[39] Dies soll ein Abbau von Glucose in der Leber verlangsamen, damit dieses eher für andere Organe zur Verfügung steht. Die Dephosphorylierung wird durch eine Phosphatase katalysiert.
Hemmstoffe
Die Enolase wird durch Fluorid inhibiert.[40] Iodacetat hemmt die Glycerinaldehyd-3-phosphat-Dehydrogenase, die Glycerinaldehyd-3-phosphat mit einem anorganischen Phosphat und unter Beteiligung von NAD+ zu 1,3-Bisphosphoglycerat oxidiert. Es modifiziert dabei eine SH-Gruppe des Enzyms, deshalb kann durch Zugabe von Mercaptanen diese Hemmung wieder aufgehoben werden.
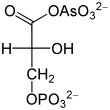 1-Arseno-3-phosphoglycerat, ein Entkoppler der Glykolyse
1-Arseno-3-phosphoglycerat, ein Entkoppler der GlykolyseArsenat, HAsO42−, ähnelt anorganischem Phosphat HPO42− und wird an seiner Stelle durch die Glycerinaldehyd-3-phosphat-Dehydrogenase umgesetzt. Dadurch entsteht aus Glycerinaldehyd-3-phosphat somit 1-Arseno-3-phosphoglycerat (vgl. Abbildung). Im Gegensatz zu 1,3-Bisphosphoglycerat ist diese Arsenatverbindung wie jedes andere Acylarsenat sehr instabil, es zerfällt zu 3-Phosphoglycerat. Infolgedessen kann die Energie der Oxidation nicht mehr durch Substratkettenphosphorylierung nutzbar gemacht werden. Es entfällt ein Schritt der ATP-Bildung in der Glykolyse, die zwar weiterhin abläuft, aber netto kein ATP mehr liefert. Da Arsenat nicht verbraucht wird, wirkt es bereits in katalytischen Mengen. Harden und Young hatten die Wirkung Arsenats auf die Glykolyse Anfang des 20. Jahrhunderts mittels Hefeextrakten demonstriert.[41]
Eintritt anderer Metabolite
Neben D-Glucose können auch andere Metabolite in der Glykolyse eintreten, sofern sie sich in eines der darin vorkommenden Zwischenprodukte umgewandelt werden können. Pentosen und Tetrosen werden dabei in der Regel durch den Pentosephosphatweg zu Glycerinaldehyd-3-phosphat beziehungsweise Fructose-6-phosphat umgewandelt und können dann weiter umgesetzt werden.
Auch die Abbauwege der Di- oder Polysacchariden münden in die Glykolyse ein. So wird zum Beispiel Saccharose in Glucose und Fructose gepalten. Wie Fructose weiter umgesetzt wird, wird weiter unten beschrieben. Beim Abbau von Milchzucker entstehen D-Glucose und D-Galactose, letzteres wird schließlich auch in Glucose umgewandelt und findet seinen Abbauweg in der Glykolyse.
Aus Vielfachzucker entstehen durch enzymatische Reaktionen einzelne Monosaccharide, die gegebenenfalls nach Isomerisierung zu Glucose-6-phosphat oder Fructose-6-phosphat direkt in den glykolytischen Abbauweg einfließen können. Ein bekanntes Beispiel ist der Speicherstoff Glykogen. Aus ihm wird durch eine Glycogenphosphorylase Glucose-1-phosphat, welches dann zu Glucose-6-phosphat isomerisiert wird.
Glycerin
Glycerin entsteht beim Abbau von Triglyceriden und kann als Vorstufe der Glykolyse beziehungsweise Gluconeogenese dienen. Eine cytosolische Glycerinkinase phosphoryliert Glycerin unter ATP-Verbrauch zu Glycerin-3-phosphat, welches dann zu Dihydroxyacetonphosphat oxidiert wird. Entweder wird dies von einer cytosolischen Glycerin-3-phosphat-Dehydrogenase (cGDH) oder einem membranständigen Isoenzym im Mitochondrium (mGDH) katalyisert. Bei ersterer wird NAD+, bei letzterer Ubichinon reduziert.
Fructose
 Eintritt von Fructose in die Glykolyse, bitte Text beachten.
Eintritt von Fructose in die Glykolyse, bitte Text beachten.
Fructose (1), Fru-1-P (2), DHAP (3), Glycerinaldehyd (4), GAP (5)
Fructokinase (FK), Aldolase B (ALD-B), Triosephosphatisomerase (TPI), Triosekinase (TK)Bei der Spaltung des Disaccharids Saccharose werden Glucose und Fructose freigesetzt. Fructose wird bei höheren Tieren in der Leber zu Fructose-1-phosphat phosphoryliert, was durch eine Fructokinase (Ketohexokinase, FK) unter ATP-Verbrauch katalysiert wird. Anschließend spaltet die Aldolase B (Fructose-1-phosphat-Aldolase, ALD-B) Fructose-1-phosphat in Dihydroxyacetonphosphat (DHAP) und Glycerinaldehyd. DHAP beziehungsweise Glycerinaldehyd werden beide in Glycerinaldehyd-3-phosphat umgewandelt. Dies katalysieren eine weiter oben beschriebene Triosephosphatisomerase (TPI) beziehungsweise eine Triosekinase (TK) unter ATP-Verbrauch. Fehlt die Fructokinase, führt dies bei höheren Tieren zu einer Fructosurie, einer autosomal-rezessiven Erbkrankheit.[42]
In anderen Organen kann auch die Hexokinase die Funktion der Fructokinase übernehmen, um Fructose zu phosphorylieren. Ihre Affinität zur Glucose gegenüber der Fructose ist aber wesentlich höher (95 % Glucose, 5 % Fructose).
Mannose
D-Mannose ist Bestandteil verschiedener Glykoproteine und Polysaccharide. Um in die Glykolyse eintreten zu können, wird Mannose zunächst durch eine Hexokinase zu Mannose-6-phosphat unter ATP-Verbrauch phosphoryliert. Dieses wird schließlich zu Fructose-6-phosphat isomerisiert, was durch Mannose-6-phosphat-Isomerase (auch Phosphomannoseisomerase, EC 5.3.1.8) katalysiert wird.
Sorbit
Sorbit kann im Polyolweg zu Glucose beziehungsweise Fructose oxidiert werden.
Galactose
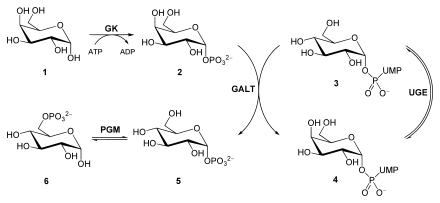 Eintritt von Galactose in die Glykolyse, bitte Text beachten.
Eintritt von Galactose in die Glykolyse, bitte Text beachten.
Galactose (1), Galactose-1-phosphat (2), UDP-Glucose (3), UDP-Galactose (4), Glu-1-P (5), Glu-6-P (6).
Galactokinase (GK), Galactose-1-phosphat-Uridyltransferase (GALT), UDP-Glucose-4-Epimerase (UGE), Phosphoglucomutase (PGM)Nach Spaltung von Milchzucker werden D-Glucose und D-Galactose freigesetzt. Um Galactose in sein C4-Epimer Glucose zu überführen, wird der Zucker zunächst durch eine Galactokinase (GK) unter Verbrauch von ATP zu Galactose-1-phosphat umgesetzt. Eine Galactose-1-phosphat-Uridyltransferase (GALT) katalysiert nun einen Austausch von UDP-gebundener Glucose mit Galactose. Hierbei entstehen Glucose-1-phosphat und UDP-Galactose. Während Glucose-1-phosphat durch eine Phosphoglucomutase (PGM) zu Glucose-6-phosphat isomerisiert wird, epimerisiert eine UDP-Glucose-4-Epimerase (UGE) UDP-Galactose zu UDP-Glucose.
Ein Defekt der Galactokinase äußert sich der Stoffwechselkrankheit Galaktosämie.
Besonderheiten bei grünen Pflanzen
Bei grünen Pflanzen gibt es einige Variationen in der Glykolyse verglichen mit der bei Tieren.[43] Diese werden im Folgenden beschrieben.
Glykolyse in den Plastiden
Es ist bekannt, dass in Pflanzen die Glykolyse nicht nur im Cytoplasma, sondern auch in den Plastiden der Zelle unabhängig voneinander betrieben wird. Nicht immer läuft sie dort jedoch vollständig ab, da häufig Enzyme der Amortisierungsphase fehlen, beispielsweise die Enolase oder die Phosphoglyceratmutase. Durch hochspezifische Translokatoren können Intermediate von einem zum anderen Zellkompartiment transportiert werden, um so alle Reaktionsschritte der Glykolyse zu vervollständigen. Im Cytosol vieler einzelliger Grünalgen fehlen die cytosolischen Enzyme für die Glykolyse, so dass diese in den Chloroplasten vollständig abläuft.
Grüne Pflanzen nutzen die Glykolyse in den Plastiden, um in der Dunkelheit oder in nicht-photosynthetischem Gewebe Stärke zu Pyruvat unter ATP- und NADH-Gewinn abzubauen. Außerdem stellen sie dadurch verschiedene Vorläufermoleküle zum Aufbau anderer Produkte bereit, beispielsweise für die Fettsäuresynthese.
Für das parallele Betreiben der Glykolyse sowohl im Cytoplasma als auch in den Plastiden werden Isoenzyme benötigt. So gibt es beispielsweise eine Pyrvatkinase, die sich im Cytoplasma befindet, als auch eine, die im Plastid die analoge Reaktion katalysiert. Alle Isoenzyme werden im Genom der Pflanze kodiert. Die plastidären Vertreter werden im Cytoplasma der Pflanzenzelle translatiert und anschließend in das Organell transportiert. Es ist noch unklar, ob die Isoenzyme durch Genduplikation aus einem Vorläufergen entstanden sind. Möglich wäre auch ein horizontaler Gentransfer aus dem Genom eines prokaryotischen Symbionten (Endosymbiontentheorie).
Rolle von Pyrophosphat
Eine weitere Besonderheit ist das Verwenden von Pyrophosphat (PPi) anstatt ATP als Phosphatdonor bei den ersten glykolytischen Reaktionen. Dies wurde ebenso in einigen Bakterien beobachtet. Normalerweise wird Pyrophosphat durch eine Pyrophosphatase (PPiase, EC 3.6.1.1) zu zwei Molekülen Phosphat hydrolysiert. Der Zweck dieser Hydrolyse ist es, biochemische Reaktionen unter physiologischen Bedingungen irreversibel zu machen. Man spricht umgangssprachlich davon, dass durch diese Hydrolyse die Reaktion auf eine Seite „gezogen“ wird. Die Erklärung dafür ist, dass die Hydrolyse exergonisch ist, dabei also Energie freigesetzt wird:
Im Cytosol der Pflanzen kommt die PPiase nicht vor, so dass dort eine Pyrophosphatkonzentration von bis zu 0,3 mmol/L entstehen kann. Die 1979 entdeckte Pyrophosphat-abhängige Phosphofructokinase (PFP, EC 2.7.1.90) nutzt das Pyrophosphat für die Phosphorylierung von Fructose-6-phosphat zu Fructose-1,6-bisphosphat. Diese Reaktion ist außerdem reversibel und könnte auch für den umgekehrten Weg, die Gluconeogenese, verwendet werden. PFP wird wie PFK-1 durch Fructose-2,6-bisphosphat reguliert.
Metabolische Vielfalt bei Phosphoenolpyruvat
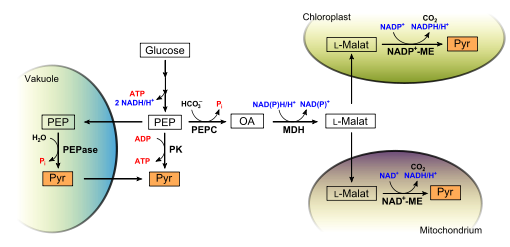 In Pflanzen kann PEP (Phosphoenolpyruvat) auf vielfältige Weise zu Pyruvat verstoffwechselt werden, bitte auch Text beachten.
In Pflanzen kann PEP (Phosphoenolpyruvat) auf vielfältige Weise zu Pyruvat verstoffwechselt werden, bitte auch Text beachten.Pflanzen nutzen PEP aus der Glykolyse in unterschiedlicher Weise (vgl. Abbildung rechts). Im klassischen, glykolytischen Abbauweg ist die Nutzung PEPs beschränkt: Eine Pyrvuatkinase (PK) nutzt PEP als Substrat zur direkten Bildung von Pyruvat. Durch Substratkettenphosphorylierung wird dabei ATP erzeugt. In Pflanzen dient PEP daürber hinaus auch als Substrat für eine PEP-Carboxykinase (PEPC), was insbesondere in Pflanzen mit C4- beziehungsweise Crassulaceen-Säurestoffwechsel wichtig ist. Aus PEP und Hydrogencarbonat entsteht hierbei Oxalacetat, ein Intermediat des Citratzyklus. Oxalacetat wird dann durch eine cytosolische Malatdehydrogenase (MDH) zu L-Malat reduziert. L-Malat wird dann in verschiedene Organellen transportiert. Im Mitochondrium kann es durch ein mitochondrielles, NAD+-abhängiges Malatenzym (ME) zu Pyruvat decarboxyliert werden. Dies ist eine Umgehungsreaktion der Pyruvatkinase, bei der aber kein ATP erzeugt wird, dafür aber bei einem Phosphatmangel nützlich ist. Denn so wird das in PEP gebundene Phosphat wieder freigesetzt und steht der Pflanze für andere Reaktionen zur Verfügung. In den Wurzeln der Erbse wurde diese Umgehung nachgewiesen.
Aus durch PEPC und MDH erzeugtem L-Malat kann ein plastidäres, NADP-abhängiges Malatenzym Pyruvat erzeugen. Pyruvat wird dort für die Fettsäuresynthese benötigt oder wird zurück ins Cytosol transportiert. Auch dadurch wird die Reaktion der Pyruvatkinase umgangen, was unter Phosphatmangel vorteilhaft ist.
Unter Phosphatmangel kann auch eine PEP-Phosphatase (PEPase, EC 3.1.3.60) wertvolles Phosphat freisetzen. Hierbei wird PEP in die Vakuole transportiert und dort von einer PEPase zu Pyruvat hydrolyisiert. Pyruvat und Phosphat werden dann ins Cytoplasma zurücktransportiert. Wenn kein Phosphatmangel vorliegt, wird die PEPase durch eine ausreichend hohe Pi-Konzentration inhibiert.
Schließlich gibt es auch eine cytosolische, phosphatunabhängige GADH (EC 1.2.1.9), die Glycerinaldehyd direkt zu 3-Phosphoglycerat oxidiert. Dabei entsteht nur NADPH, aber kein ATP.
Regulation
Bei der Regulation der an der Glykolyse beteiligten Enzyme gibt es einige wichtige Unterschiede zu der bei Tieren.[43] So ist PEP ein besonderer allosterischer Effektor, das im Gegensatz zum Ablauf in Tieren die PFK inhibieren kann. Fructose-1,6-bisphosphat dagegen kann nicht die Pyruvatkinase aktivieren. Während Fructose-2,6-bisphosphat in Tieren die PFK aktiviert, geschieht nichts dergleichen in Pflanzen.
Damit wird die Glykolyse in grünen Pflanzen in erster Linie durch die Aktivitäten der Pyruvatkinase und PEP-Carboxykinase reguliert, in zweiter Linie durch die PFK-1 beziehungsweise PFP. Bei Tieren ist es prinzipiell umgekehrt.
Glykolyse bei Archaeen
Bei den zuckerabbauenden Archaeen werden Kohlenhydrate auf unterschiedliche Weise abgebaut. In hyperthermophilen und thermophilen Aerobiern, beispielsweise Thermoplasma acidophilum oder Sulfolobus solfataricus, wird Glucose über eine Variante des Entner-Doudoroff-Weg (ED-Weg) zu Pyrvuat umgesetzt. Dagegen verwerten hyperthermophile, gärende Anaerobier wie Pyrococcus furiosus, Desulfurococcus amylolyticus, Pyrobaculum aerophilum (ein Mikroaerobier), der Sulfatreduzierer Archaeoglobus fulgidus sowie Vertreter der Gattung Thermococcus Kohlenhydrate in einen modifizierten EMP-Weg.[45]
Die darin vorkommenden Metabolite gleichen zwar denen der Glykolyse von Eukaryoten und Bakterien, jedoch werden hierfür Enzyme verwendet, die keine Ähnlichkeiten mit denen von Bakterien oder Eukaryoten aufweisen. So findet man in Archaeen ADP- anstatt ATP-abhängige Kinasen, beispielsweise die Glucokinase (EC 2.7.1.147) oder die Phosphofructokinase (EC 2.7.1.146). Im Gegensatz zu Bakterien verfügen sie nicht über ein PEP-abhängiges Zuckertransportsystem. Der letzte Schritt in der Glykolyse, die Umsetzung von PEP zu Pyruvat, kann neben der PK auch durch eine Pyruvat-Phosphat-Dikinase (PPDK, EC 2.7.9.1) erfolgen. Dieses Enzym katalysiert die reversible Umwandlung von PEP, AMP und PPi zu Pyruvat, ATP und Pi, wenngleich in Thermoproteus tenax die Bildung von Pyruvat bevorzugt wird.[46]T. tenax ist ein anaerob lebendes, fakultatives, heterotrophes Archeon der Abteilung Crenarchaeota. PPDK wurde auch in Bakterien und Eukaryoten nachgewiesen.
Ein wichtiger Unterschied besteht bei der Oxidation von Glycerinaldehyd-3-phosphat. Dieses wird entweder von einer NAD(P)+-abhängigen (GAPN) oder einer Ferredoxin-abhängigen Dehydrogenase (GAPOR) direkt zu 3-Phosophoglycerat oxidiert, ohne aber dabei anorganisches Phosphat einzubauen.[22] Daher wird bei diesem Schritt kein ATP durch Substratkettenphosphorylierung gebildet, so dass bei den meisten Archaeen durch diese modifizierte Glykolyse formal kein ATP gewonnen wird. Der am besten untersuchte Glykolysestoffwechselweg in Archaeen ist der von P. furiosus. Dort beträgt die Nettoreaktion:
Ein weiterer wichtiger Unterschied ist die fehlende allosterische Regulation der Schlüsselenzyme mit den Effektoren, die für Bakterien beziehungsweise Eukaryoten weiter oben beschrieben wurde. In T. tenax gibt es zumindest Hinweise darauf, dass die GAPN durch AMP, Glucose-1-phosphat, Fructose-6-phosphat, Fructose-1-phosphat, ADP und Ribose-5-phosphat allosterisch aktiviert wird, während NAD(P)H, NADP+ und ATP das Enzym hemmen. Möglicherweise findet auch eine Regulation auf Ebene der Transkription statt, wie bei der in T. tenax vorkommenden GAPDH.[47]
Evolution
Die Glykolyse ist in den meisten Bakterien und Eukaryoten vertreten, in etwas abgewandelter Form auch bei Archaeen und hyperthermophilen Bakterien. Dies lässt darauf schließen, dass die Glykolyse sich sehr früh im Laufe der Evolution etabliert hatte und bereits in den ersten Organismen vertreten war. Durch phylogenetische Vergleiche mit thermophilen und hyperthermophilen Mikroorganismen vermutet man, dass der EMP-Weg nach dem ED-Weg entstanden ist. Außerdem lag wahrscheinlich die ursprüngliche Bedeutung des EMP-Weges nicht im Abbau von Kohlenhydraten, sondern er lief umgekehrt als Gluconeogenese zum Aufbau von Glucose ab.[48][49] Dies stützt auch die Theorie, dass Stoffwechselwege für den Aufbau von Kohlenhydraten in der Evolution früher aufgetreten sind als solche, die Kohlenhydrate abbauen; so ist die „heutige“ Gluconeogenese bei den Organismen aller drei Domänen weiter verbreitet als die Glykolyse.
Die Enzyme des unteren Zweiges der Glykolyse (Amortisierungsphase) katalysieren größtenteils Reaktionen, die reversibel ablaufen und am höchsten konserviert sind.[22] Außerdem kommen sie auch in dem phylogenetisch älteren ED-Weg vor. Wahrscheinlich waren sie – wie auch andere Stoffwechselwege – schon vor der Auftrennung der drei Domänen der Lebewesen vorhanden und zählen daher zu den ältesten Enzymen. Wegen ihrer essentiellen Bedeutung konnten sie weder verloren gehen noch im Laufe der Zeit durch andere Enzyme ersetzt werden. Die Glycerinaldehyd-3-phosphat-Dehydrogenase ist unter allen glykolytischen Enzymen am höchsten konserviert, lediglich 3 % der katalytischen Domäne haben sich in 100 Millionen Jahren verändert.
Der obere Zweig der Glykolyse (Vorbereitungsphase) hat sich vermutlich später etabliert. Während die darin involvierten Enzyme in Bakterien und Eukaryoten hohe Homologien aufweisen, sind die in Archaeen vorkommenden Enzyme dagegen einzigartig. Es wird noch diskutiert, ob ursprünglich Enzyme für den ersten Teil der Glykolyse bei Archaeen verloren gingen und erst später durch horizontalen Gentransfer wieder eingeführt wurden. Alternativ könnten auch Enzyme mit ähnlicher Funktionen für die ursprüngliche Glykolyse herangezogen worden sein, welche danach starken Modifikationen und Sequenzänderungen unterlagen. In beiden Fällen könnte dies erklären, warum sich die glykolytischen Enzyme in Archaeen so sehr von denen anderer Organismen unterscheiden.
Literatur
- Geoffrey Zubay: Biochemie. Mcgraw-Hill Professional; 4. Auflage 1999; ISBN 3-89028-701-8; S. 293ff.
- Donald Voet und Judith G. Voet: Biochemie. Wiley-VCH 1994; ISBN 3-527-29249-7; S. 420ff.
- Jeremy M. Berg, John L. Tymoczko, Lubert Stryer: Biochemie. 6 Auflage. Spektrum Akademischer Verlag, Heidelberg 2007; ISBN 978-3-8274-1800-5; S. 486ff.
- H. Robert Horton, Laurence A. Moran, K. Gray Scrimgeour, Marc D. Perry, J. David Rawn und Carsten Biele (Übersetzer): Biochemie. Pearson Studium; 4. aktualisierte Auflage 2008; ISBN 978-3-8273-7312-0; S. 442ff.
- Reginald Garrett und Charles M. Grisham: Biochemistry. (International Student Edition). Cengage Learning Services; 4. Auflage 2009; ISBN 978-0-495-11464-2; S. 535–562
- Nelson, David L., Cox, Michael M., Lehninger, Albert L. [Begr.]: Lehninger Biochemie. Springer, Berlin; 4., vollst. überarb. u. erw. Auflage 2009; ISBN 978-3-540-68637-8; S. 607–730
- Ronimus, RS. und Morgan, HW. (2003): Distribution and phylogenies of enzymes of the Embden-Meyerhof-Parnas pathway from archaea and hyperthermophilic bacteria support a gluconeogenic origin of metabolism. In: Archaea 1(3); 199–221; PMID 15803666; PDF (freier Volltextzugriff, engl.)
- Plaxton, WC. (1996): The organization and regulation of plant glycolysis. In: Annu Rev Plant Physiol Plant Mol Biol. 47; 185–214; PMID 15012287; doi:10.1146/annurev.arplant.47.1.185
- Dandekar T, Schuster S, Snel B, Huynen M, Bork P: Pathway alignment: application to the comparative analysis of glycolytic enzymes. In: Biochem. J.. 343 Pt 1, Oktober 1999, S. 115–24. PMID 10493919. Volltext bei PMC: 1220531.
Einzelnachweise
- ↑ Cagniard-Latour, C. (1837): Mémoire sur la fermentation vineuse. In: Annales de chimie et de physique. Bd. 68, 1837, S. 206–222
- ↑ Theodor Schwann (1837): Vorläufige Mitteilung, betreffend Versuche über die Weingährung und Fäulnis. In: Annalen der Physik und Chemie. Bd. 41, S. 184–193
- ↑ Kützing, F. T. (1837): Microscopische Untersuchungen über die Hefe und Essigmutter, nebst mehreren andern dazu gehörigen vegetabilischen Gebilden. In: Journ. prakt. Chem. 11, S. 385–409
- ↑ Racker, E. (1974): History of the Pasteur effect and its pathobiology. In: Mol Cell Biochem. 5(1–2); 17–23; PMID 4279327; doi:10.1007/BF01874168
- ↑ Liebig, J.(1839): Ueber die Erscheinungen der Gährung, Fäulniß und Verwesung und ihre Ursachen. In: Annalen der Pharmacie (Heidelberg) 30, S. 250–287
- ↑ Pasteur, L. (1860): Memoire sur la fermentaçion alcoolique. In: Annales de chimie et de physique 58; 323-426
- ↑ Traube, M. (1858). Ann. Phys. Chem. (Poggendorff) 103; 331–344
- ↑ Buchner, E. (1897): Alkoholische Gärung ohne Hefezellen (Vorläufige Mitteilung). In: Ber. Dt. Chem. Ges. 30, 117-124; Online
- ↑ Athel Cornish-Bowden: New beer in an old bottle: Eduard Buchner and the growth of biochemical knowledge. ISBN 84-370-3328-4, S. 60
- ↑ Harden, A. und Young, WJ. (1906): The Alcoholic Ferment of Yeast-Juice. In: Proc. R. Soc. Lond. B 77(519); 405–420; doi:10.1098/rspb.1906.0029
- ↑ Harden, A. und Young, WJ. (1908): The Alcoholic Ferment of Yeast-Juice. Part III.-The Function of Phosphates in the Fermentation of Glucose by Yeast-Juice. In: Proc. R. Soc. Lond. B 80(540); 405–420; doi:10.1098/rspb.1908.0029
- ↑ Donald Voet, Judith G. Voet, Alfred Maelicke (Hsrg.), Werner Müller-Esterl (Hrsg.): Biochemie. Wiley-VCH 1992. ISBN 3-527-28242-4; S. 421f.
- ↑ Kresge N., Simoni, RD. und Hill, RL. (2005): Otto Fritz Meyerhof and the elucidation of the glycolytic pathway. In: J Biol Chem. 280(4):e3; PMID 15665335; PDF (freier Volltextzugriff, engl.)
- ↑ Albert L. Lehninger, David L. Nelson und Michael M. Cox: Lehninger Biochemie. 3., vollst. überarb. u. erw. Auflage. Springer, Berlin 2009, ISBN 978-3-540-41813-9; S. 584ff.
- ↑ Donald Voet, Judith G. Voet, Alfred Maelicke (Hsrg.), Werner Müller-Esterl (Hrsg.): Biochemie. Wiley-VCH, 1992, ISBN 3-527-28242-4; S. 444
- ↑ Hans W. Heldt, Birgit Piechulla: Pflanzenbiochemie. 4. Auflage. Spektrum Akademischer Verlag, Heidelberg 2008, ISBN 978-3-8274-1961-3, S. 334
- ↑ Reginald Garrett und Charles M. Grisham: Biochemistry. (International Student Edition). 4. Auflage. Cengage Learning Services, 2009, ISBN 978-0-495-11464-2, S. 535
- ↑ Hans W. Heldt, Birgit Piechulla: Pflanzenbiochemie. 4. Auflage. Spektrum Akademischer Verlag, Heidelberg 2008, ISBN 978-3-8274-1961-3, S. 334.
- ↑ Jeremy M. Berg, John L. Tymoczko, Lubert Stryer: Biochemie. 6 Auflage. Spektrum Akademischer Verlag, Heidelberg 2007, ISBN 978-3-8274-1800-5, S. 764
- ↑ Caroline Bowsher, Martin W. Steer und Alyson K. Tobin: Plant Biochemistry. Garland Pub, New York, NY 2008, ISBN 978-0-8153-4121-5; S. 145
- ↑ H. Robert Horton, Laurence A. Moran, K. Gray Scrimgeour, Marc D. Perry, J. David Rawn und Carsten Biele (Übersetzer): Biochemie. Pearson Studium; 4. aktualisierte Auflage 2008; ISBN 978-3-8273-7312-0; S. 448
- ↑ a b c Ronimus, RS und Morgan, HW. (2003): Distribution and phylogenies of enzymes of the Embden-Meyerhof-Parnas pathway from archaea and hyperthermophilic bacteria support a gluconeogenic origin of metabolism. In: Archaea. 1(3); 199–221; PMID 15803666; PDF (freier Volltextzugriff, engl.)
- ↑ a b Caroline Bowsher, Martin W. Steer und Alyson K. Tobin: Plant Biochemistry. Garland Pub, New York, NY 2008, ISBN 978-0-8153-4121-5; S. 148
- ↑ David Nelson, Michael Cox: Lehninger Biochemie. Springer, Berlin; 4., vollst. überarb. u. erw. Auflage 2008; ISBN 978-3-540-68637-8; S. 713
- ↑ Caroline Bowsher, Martin W. Steer und Alyson K. Tobin: Plant Biochemistry. Garland Pub, New York, NY 2008, ISBN 978-0-8153-4121-5; S. 150
- ↑ a b c d Reginald Garrett und Charles M. Grisham: Biochemistry. (International Student Edition). 4. Auflage 2009, Cengage Learning Services, ISBN 978-0-495-11464-2, S. 538
- ↑ a b David L. Nelson und Michael M. Cox: Lehninger Principles of Biochemistry. Palgrave Macmillan; 5. Auflage 2008; ISBN 978-0-7167-7108-1; S. 553
- ↑ Reginald Garrett und Charles M. Grisham: Biochemistry. (International Student Edition). 4. Auflage, Cengage Learning Services, Australien 2009, ISBN 978-0-495-11464-2, S. 539
- ↑ S. Minakami, H. Yoshikawa: Studies on erythrocyte glycolysis. II. Free energy changes and rate limitings steps in erythrocyte glycolysis. In: J. Biochem. Bd. 59(2) 1966, S. 139–144; PMID 4223318
- ↑ S. Minakami, H. Yoshikawa: Thermodynamic considerations on erythrocyte glycolysis. In: Biochem. Biophys. Res. Commun. Bd. 18(3) 1965; S. 345–349; PMID 14300746; doi:10.1016/0006-291X(65)90711-4
- ↑ Reginald Garrett und Charles M. Grisham: Biochemistry. (International Student Edition). 4. Auflage, Cengage Learning Services, Australien 2009, ISBN 978-0-495-11464-2, S. 554
- ↑ Athel Cornish-Bowden: New beer in an old bottle: Eduard Buchner and the growth of biochemical knowledge. Universitat de Valencia, Valencia 1997, ISBN 84-370-3328-4, S. 148
- ↑ Reginald Garrett und Charles M. Grisham: Biochemistry. (International Student Edition). 4. Auflage 2009, Cengage Learning Services, ISBN 978-0-495-11464-2, S. 537
- ↑ Newsholme, EA. et al. (1972): The activities of fructose diphosphatase in flight muscles from the bumble-bee and the role of this enzyme in heat generation. In: Biochem J. 128(1); 89–97; PMID 4343671; PDF (freier Volltextzugriff, engl.)
- ↑ a b Reginald Garrett und Charles M. Grisham: Biochemistry. (International Student Edition). Cengage Learning Services; 4. Auflage 2009; ISBN 978-0-495-11464-2; S. 539f.
- ↑ a b c David Nelson, Michael Cox: Lehninger Biochemie. Springer, Berlin; 4., vollst. überarb. u. erw. Auflage 2008; ISBN 978-3-540-68637-8; S. 773ff.
- ↑ Reginald Garrett und Charles M. Grisham: Biochemistry. (International Student Edition). Cengage Learning Services; 4. Auflage 2009; ISBN 978-0-495-11464-2; S. 542f.
- ↑ a b c H. Robert Horton, Laurence A. Moran, K. Gray Scrimgeour, Marc D. Perry, J. David Rawn und Carsten Biele (Übersetzer): Biochemie. Pearson Studium; 4. aktualisierte Auflage 2008; ISBN 978-3-8273-7312-0; S. 468
- ↑ H. Robert Horton, Laurence A. Moran, K. Gray Scrimgeour, Marc D. Perry, J. David Rawn und Carsten Biele (Übersetzer): Biochemie. Pearson Studium; 4. aktualisierte Auflage 2008; ISBN 978-3-8273-7312-0; S. 470
- ↑ Todd A. Swanson, Sandra I. Kim und Marc J. Glucksman: BRS Biochemistry, Molecular Biology, and Genetics. Lippincott Raven; 5. Auflage 2010; ISBN 978-0-7817-9875-4; S. 65
- ↑ Harden, A. und Young, WJ (1911): The Alcoholic Ferment of Yeast-Juice. Part VI.-The Influence of Arsenates and Arsenites on the Fermentation of the Sugars by Yeast-Juice. In: Proc. R. Soc. Lond. B 83(566); 451–475; doi:10.1098/rspb.1911.0028
- ↑ Todd A. Swanson, Sandra I. Kim und Marc J. Glucksman: BRS Biochemistry, Molecular Biology, and Genetics. Lippincott Raven; 5. Auflage 2010; ISBN 978-0-7817-9875-4; S. 123
- ↑ a b Plaxton, WC. (1996): The organization and regulation of plant glycolysis. In: Annu Rev Plant Physiol Plant Mol Biol. 47; 185–214; PMID 15012287; doi:10.1146/annurev.arplant.47.1.185
- ↑ Rudolf K. Thauer, Kurt Jungermann, Karl Decker: Energy conservation in chemotrophic anaerobic bacteria. In: Bacteriological Reviews. Bd. 41, Nr. 1, 1977, S. 101.
- ↑ Siebers, B. und Schönheit, P. (2005): Unusual pathways and enzymes of central carbohydrate metabolism in Archaea. In: Curr Opin Microbiol. 8(6); 695–705; PMID 16256419; doi:10.1016/j.mib.2005.10.014
- ↑ Zaparty, M. et al. (2008): The central carbohydrate metabolism of the hyperthermophilic crenarchaeote Thermoproteus tenax: pathways and insights into their regulation. In: Arch Microbiol. 190(3); 231–245; PMID 18491075; doi:10.1007/s00203-008-0375-5
- ↑ Verhees, CH. et al. (2003): The unique features of glycolytic pathways in Archaea. In: Biochem J. 375(Pt 2); 231–246; PMID 12921536; PDF (freier Volltextzugriff, engl.)
- ↑ A. H. Romano, T. Conway: Evolution of carbohydrate metabolic pathways. In: Res. Microbiol. Bd. 147 (6–7), 1996, S. 448–455, PMID 9084754; doi:10.1016/0923-2508(96)83998-2
- ↑ Ronimus, RS. und Morgan, HW. (2003): Distribution and phylogenies of enzymes of the Embden-Meyerhof-Parnas pathway from archaea and hyperthermophilic bacteria support a gluconeogenic origin of metabolism. In: Archaea 1(3); 199–221; PMID 15803666; PDF (freier Volltextzugriff, engl.)
Weblinks
 Wikibooks: Biochemie und Pathobiochemie: Glycolyse – Lern- und Lehrmaterialien
Wikibooks: Biochemie und Pathobiochemie: Glycolyse – Lern- und Lehrmaterialien Commons: Glykolyse – Sammlung von Bildern, Videos und Audiodateien Commons: Stoffwechselwege der Glykolyse – Sammlung von Bildern, Videos und Audiodateien Commons: Enzyme der Glykolyse – Sammlung von Bildern, Videos und Audiodateien
Commons: Glykolyse – Sammlung von Bildern, Videos und Audiodateien Commons: Stoffwechselwege der Glykolyse – Sammlung von Bildern, Videos und Audiodateien Commons: Enzyme der Glykolyse – Sammlung von Bildern, Videos und Audiodateien Wiktionary: Glykolyse – Bedeutungserklärungen, Wortherkunft, Synonyme, Übersetzungen
Wiktionary: Glykolyse – Bedeutungserklärungen, Wortherkunft, Synonyme, Übersetzungen- Die Glycolyse auf der Homepage von Ulrich Helmich
- Audio-Beitrag zum Thema Glykolyse
- Jennifer McDowall/Interpro: Protein Of The Month: Enzymes of Glycolysis. (engl.)


Dieser Artikel wurde am 21. Juli 2010 in dieser Version in die Liste der exzellenten Artikel aufgenommen. Kategorien:- Biologischer Prozess
- Wikipedia:Exzellent
- Biochemische Reaktion
- Stoffwechselweg


































Wikimedia Foundation.